Full Text
Introduction
Atypical teratoid rhabdoid tumour is a rare and very aggressive malignant embryonal tumour of central nervous system (CNS) seen in infancy and early childhood. The exact incidence of childhood CNS AT/RT is difficult to determine because this tumor as a separate entity has been widely recognized only the last decade. Historically these tumors, also known as malignant rhabdoid tumour, were commonly mistaken as primitive neuroectodermal tumors because of certain similar morphological features on histopathological examination [1, 2].
The age at presentation is usually less than 2 years. It has also been rarely reported to occur in older children and adults [4, 5]. Here we present a case of young adult with a suprasellar region atypical teratoid/ rhabdoid tumor, an unusual adult cancer presenting in an uncommon location, with a brief review of literature of this rarely reported disease in adult patients with an emphasis on treatment and positive outcomes with addition of radiotherapy in the multimodality management.
Case report
A 20 year old male medical student presented with history of blurring of vision of 4 months duration. Patient also had tingling and numbeness and weakness of right hand of 2 weeks duration. There was no history of headache, vomiting or seizures. On examination his higher mental functions were normal, there was weakness of right thenar muscles. Routine blood investigations, serum chemistry and hormonal assays were within normal limits. MRI brain revealed suprasellar lesion which was enhancing after contrast administration. Lesion was causing compression over optic chiasma.
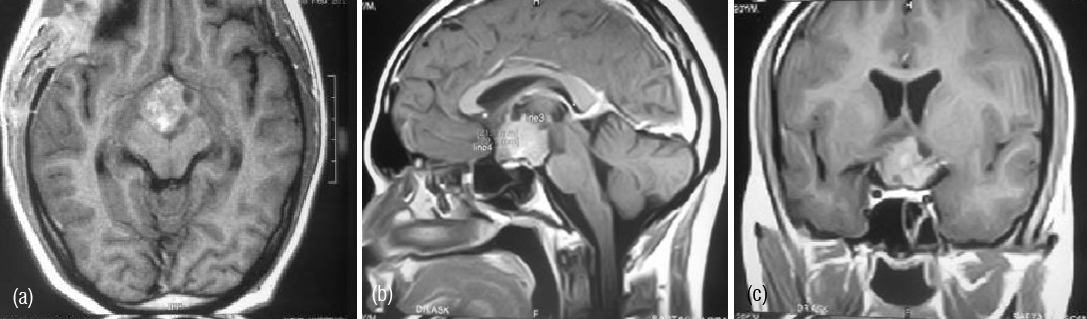
Figure 1a,b,c: MRI -contrast enhancing mass lesion in sellar and suprasellar region.
Patient underwent right pterional craniotomy and subtotal resection of suprasellar lesion. Per-operatively there was grayish firm mildly vascular lesion with calcification in suprasellar region with compression of optic apparatus. Post operative histopathology showed moderately cellular neoplasm with nuclear atypia, hyperchromasia. Several cells had rhabdoid like morphology. Foci of micronecrosis were present. It was reported as atypical cellular tumour with rhabdoid morphology.

Figure 2a,b: Cellular neoplasm with multiple rhabdoid cells.
Immunohistochemistry showed glial fibrillary acidic protein (GFAP), desmin & chromogranin negativity, synaptophysin had faint cytoplasmic positivity, epithelial membrane antigen showed focal positivity in clear cells but was negative in rhabdoid cells. Mib-1 index was positive in upto 25% of cells. INI -1 staining was lost in tumour cells but was retained in endothelial cells.
Post operatively, MRI brain and screening of entire spine was done which revealed residual lesion in suprasellar region and interpeduncular cistern. Another intramedullary lesion at C3-C4 level was detected causing focal cord expansion and displacement of cord anteriorly suggesting metastasis.
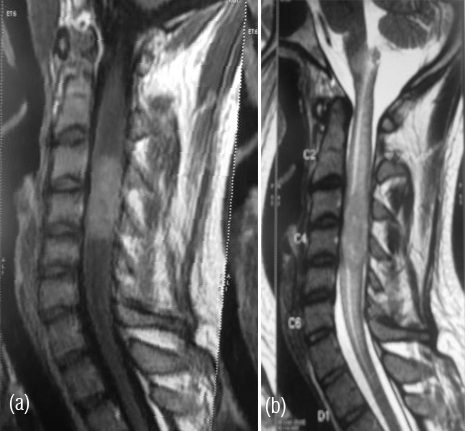
Figure 3a,b: MR spine – An elongated cylindroid space occupying lesion in cervical cord.
Metastatic work up also included chest radiograph, cerebrospinal fluid (CSF) cytology for malignant cells, radionuclide bone scan, bone marrow examination and ultrasound sonography (USG) abdomen, which did not reveal any other metastatic deposits outside the CNS.
Patient was treated with adjuvant radiation therapy to entire craniospinal axis + a focal boosts to residual suprasellar and spinal lesions followed by adjuvant chemotherapy and is disease free at 6 months follow-up.
Discussion
Beckwith and Palmer in 1978 first coined the term ‘rhabdoid tumor’ to describe a histological variant of Wilm’s tumor seen usually in infants which had extremely poor prognosis [6]. Atypical teratoid/ rhabdoid tumor was first described as a distinct entity in 1987 [1] and is now included in WHO classification of brain tumours (1993) [7].
Atypical teratoid rhabdoid tumor (AT/ RT) is a rare tumor usually diagnosed in infancy and early childhood. Although usually a brain tumor, AT/ RT can occur in other areas of the central nervous system including the spinal cord. About 60% occur in the posterior cranial fossa. In a review estimated the occurrence in various sub sites of CNS; 52% were seen in posterior fossa, 39% in supratentorial location, 5% pineal and 2% in spinal region.
An estimated 3% of pediatric brain tumors are AT/RTs; although the exact incidence of childhood CNS AT/RT is difficult to determine because the tumor has been widely recognized only in the last decade. In two recent North American based prospective studies performed by the Children’s Cancer Group and the Pediatric Oncology Group for children aged 3 years or younger at diagnosis, reported that approximately 10% of children had AT/RTs [3]. As compared to other CNS tumors, males are more affected than females (ratio 1.6:1).
Histology
Histologically, classic AT/RT is morphologically heterogeneous tumor. It typically contains many different types of cells including the rhabdoid cells, large spindle cell, epithelial and mesenchymal cells and areas resembling primitive neuroectodermal tumor (PNET). As much as 70% of the tumor may be made up of PNET-like cells (small round blue cells).
Immunohistochemical staining for epithelial markers (cytokeratin or epithelial membrane antigen), glial fibrillary acidic protein, synaptophysin (or neurofilament), and smooth muscle (desmin) may help to identify the heterogeneity of differentiation, but will vary depending on the cellular composition [8]. Rhabdoid cells, if present, will express vimentin, epithelial membrane antigen, and smooth muscle actin.
It is a rapidly growing tumor and usually has MIB-1 labeling index of 50% to 100% [7]. Immunohistochemistry for INI1 protein is useful in establishing the diagnosis of AT/ RT. A loss of INI1 staining is noted in neoplastic cells, but is retained in non-neoplastic cells [8, 9].
Genetics
AT/ RT is the first primary pediatric brain tumor for which a candidate tumor suppressor gene, SMARCB1 (also known as INI1 and hSNF5), has been identified [10]. SMARCB1 has been found to be abnormal in the majority of rhabdoid tumors, including CNS, renal, and other extrarenal rhabdoid malignancies [11]. The chromosome 22 area contains the hSNF5/ INI1 gene that appears to function as a classic tumor suppressor gene. This mutation is viewed as the "first hit" which predisposes children to malignancies. Identification of INI1 as a tumor suppressor has facilitated accurate diagnosis of rhabdoid tumors.
Medulloblastmas/ PNETs may possibly be differentiated cytogenetically from AT/RTs. Chromosomal deletions of 17p are relatively common with medulloblastoma and abnormalities of 22q11 are not seen. On the other hand, chromosomal 22 deletions are very common in AT/RTs.
However, it should be noted that this mutation of chromosome 22 in AT/ RT is not present in 100% of cases. Therefore, if the mutation is not present in an otherwise classic AT/ RT immunohistochemical and morphologic pattern then the diagnosis still remains an AT/ RT.
Treatment
There is no established standard treatment for atypical teratoid/ rhabdoid tumor (AT/ RT). Given the highly aggressive nature of the tumor, most patients have been treated with intensive multimodal therapy. The young age of the majority of patients limits the extent of treatment, particularly radiation. Initial surgery includes debulking of tumour and providing tissue sample for accurate diagnosis. Data from the AT/ RT Registry suggest that patients who undergo complete resection may have a longer median survival, although complete surgical resection is difficult in view of invasive nature of the tumor.
The traditional practice of treatment for infants has been to use multiagent chemotherapy and to defer radiation therapy until a child is older than three years in view of risk of severe neurocognitive deficits associated with the use of whole craniospinal axis irradiation.
But survival outcomes have been very poor when treated with standard chemotherapy alone. The Children’s Cancer Group reported a 2-year event-free survival (EFS) of only 14% in children younger than 3 years treated with multiagent chemotherapy [12].
Radiation therapy has definite positive impact on survival for AT/ RT patients. Of the 42 patients in the AT/ RT registry, 13 patients (31%) received radiation therapy in addition to chemotherapy as part of their primary therapy [13]. Median survival of patients receiving radiation was 48 months, while the median survival of all patients on the registry was only 16.75 months.
A retrospective survey from St. Jude Children's Hospital of 36 patients from 1984 to 2003 showed that the two-year event-free survival (EFS) for children under three was 11%, the overall survival (OS) rate was 17%. For children aged 3 years or older the EFS was 78% and the OS 89%.
Outcomes of AT/ RT are so poor that some protocols recommend upfront radiation therapy in spite of young age. In older children and adults, surgical resection is typically followed by Cranio-spinal axis irradiation and then adjuvant multiagent chemotherapy.
Prognosis
Patients with metastasis, larger tumors, tumors that could not be fully removed, or tumor recurrence, and who were younger than 36 months, had the worst outcomes. Younger patients are more likely to have disseminated disease at diagnosis and tend to develop disease progression and/ or recurrence with higher frequency and earlier in the course of therapy than older patients.
Conclusion
Atypical teratoid/ rhabdoid tumor is frequently confused with primitive neuroectodermal tumor based on histopathology evaluation because of their similar features. Awareness of atypical teratoid/ rhabdoid tumor is important in making the correct diagnosis of this uncommon but under diagnosed entity. All patients with childhood AT/ RT should have magnetic resonance imaging of the brain and spine and cerebrospinal fluid examination in order to establish the extent of spread. Ultrasound or CT scan abdomen should be done to detect synchronous renal tumors. There is no method to reliably distinguish AT/ RT from other malignant brain tumors based on clinical history or radiographic evaluation alone. Embryonal tumors, especially in those younger than 3 years, should undergo immunostaining for loss of INI1protein expression to confirm the diagnosis. Treatment approach should be aggressive multimodality therapy consisting of maximum safe resection followed by adjuvant entire neuraxis radiation and multidrug chemotherapy.
Acknowledgement
Acknowledgements are due to department of Radiology and Imaging, KIMS, Secunderabad.
Conflict of Interest
The authors wish to express that they have no conflict of interest.
References
1. Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996; 85(1):56-65.
2 Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998; 22(9):1083-1092.
3. Packer RJ, Biegel JA, Blaney S, Finlay J, Geyer JR, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol. 2002; 24(5):337-342.
4. Lutterbach J, Liegibel J, Koch D, Madlinger A, Frommhold H, et al. Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol. 2001; 52(1):49-56.
5. Pimentel J, Silva R, Pimentel T. Primary malignant rhabdoid tumors of the central nervous system: considerations about two cases of adulthood presentation. J Neurooncol. 2003; 61(2):121-126
6. Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer 1978; 41(5):1937-1948.
7. Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002; 61(3):215-225;
8. Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011; 56(1):7-15.
9. Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011; 35(6):933-935
10. McLendon RE, Adekunle A, Rajaram V, Koçak M, Blaney SM. Embryonal central nervous system neoplasms arising in infants and young children: a pediatric brain tumor consortium study. Arch Pathol Lab Med. 2011; 135(8):984-993.
11. Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, et al. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002; 8(11):3461-3467.
12. Geyer JR, Sposto R, Jennings M, Boyett JM, Axtell RA, et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol. 2005; 23(30):7621-7631.
13. Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004; 22(14):2877-2884.