Full Text
In radiotherapy, a number of factors can influence the responses of tumours and surrounding normal tissues. These factors may depend on the patient, the tumour, or the normal tissue concerned. One important prognostic factor is the susceptibility of different cell types to undergo cellular demise after irradiation. Cell death can occur via several major pathways [1-3]. In some modes of cell death, particularly apoptosis, mitochondrial membrane integrity plays a significant role. Translocation of the pro-apoptotic Bax or its homologue Bak onto mitochondria facilitates the release of cytochrome c to mediate subsequent cell death [4, 5]. Therefore, identification of mitochondria-mediated cell death as a major mode of cellular demise following radiation treatment might provide an insight into the possibility of using Bax or Bak expression as a prognostic indicator.
Although the tumour suppressor gene p53 is known to play a key role in the translocation of Bax or Bak onto mitochondria to mediate in apoptotic cell death, its functionality and wild- type status are not required for cells to undergo Bax- or Bak-mediated apoptosis [6,7]. In fact, the translocation of Bax to mitochondria in response to exposure to cytotoxic agents has been demonstrated in cells lacking functional p53 [8, 9]. Given that most tumours have mutated or dysfunctional p53, it is conceivable that the expression of Bax and/or Bak has a pivotal role in cellular response to radiation insult. The importance of Bax as a prognostic marker in radiotherapy has been highlighted by several reports [10-14]. There is evidence to show that in the absence of Bax functionality, apoptotic cell death may be mediated by Bak [15].
This study was designed to investigate whether cell death in the Bax-negative human prostate cancer cell line, DU145, would preferentially occur via the Bak-mediation after photon and neutron irradiation. The role of radiation-induced dissipation of mitochondrial membrane potential in radiosensitivity on the basis of clonogenic cell survival is further discussed.
Materials and methods
Cell lines and culture maintenance
Human prostate cancer cells (DU145) were used in this study. The tumour suppressor gene p53, which plays a significant role in cellular response to radiation insult, is known to be mutated in this cell line [16]. Also, these cells are Bax-negative but do express the pro-apoptotic Bax homologue Bak [17, 18]. Cells were cultured in minimum essential medium as monolayers at 37C in a 5% carbon dioxide humidified atmosphere in air. Growth medium was supplemented with 10 g/ml streptomycin and 10 U/ml penicillin (Highveld Biological, South Africa), and contained 10% heat-inactivated foetal bovine serum (Sigma-Aldrich, USA).
Mitochondrial dissipation assay
Cells in exponential growth were trypsinised, seeded into 25-cm2 cell culture flasks and incubated for 3 h to attach. Cultures were then irradiated to graded doses in the ranges 0-6 and 0-4 Gy with low linear energy transfer (LET) 60Co -rays (~0.5 keV/µm) and high LET p(66)/Be neutrons (~30 keV/µm), respectively. Photon irradiation was performed with an upward vertical beam from a 60Co g-ray irradiator at a dose rate of 0.203±0.004 Gy/min (range: 0.196-0.208 Gy/min). Samples were placed on a 6-mm perspex plate acting as a build-up material. A 5-cm thick perspex block was used for backscattering. The field used was 30x30 cm2. For the accelerator produced p(66)/Be neutron exposure, a vertical beam facing downward was employed at a dose rate of 0.42 Gy/min. The cell samples were placed on a 15-cm thick perspex backscatter block, and build-up material consisted of a 20-mm polyethylene block. The field used was 29x29 cm2. Cell cultures were irradiated at room temperature (22°C) and were then re-incubated for 20 h prior to the evaluation of mitochondrial dissipation.
Changes in mitochondrial membrane potential were assessed using the DePsipherTM Kit per manufacturer’s instructions (Assay Designs, USA; cat # 900-167). Briefly, cells were trypsinised, counted, and centrifuged to produce a pellet of ~106 cells per sample. Each pellet was resuspended in 1 ml of pre-warmed DePsipherTM staining solution in reaction buffer, premixed with stabiliser solution (final concentration of 2%), and incubated at 37C in a 5% carbon dioxide humidified atmosphere in air for 20 minutes. The stained cells were then centrifuged at 200 g for 5 minutes, and the pellet was resuspended in 1 ml of stabiliser- containing reaction buffer. The DePsipher™ dye is a lipophilic cation and aggregates on polarised mitochondrial membranes of healthy cells to form an orange-red fluorescent compound. If the membrane potential is compromised, the dye cannot access the transmembrane space and remains monomeric and fluoresces green. The cells were observed immediately by fluorescence microscopy, using a Zeiss Axioskop 50 microscope with a band-pass filter for fluorescein and rhodamine. At least 1 000 cells per sample and dose point were evaluated. Three independent experiments were performed for each irradiation dose. The frequency of cells with compromised mitochondrial membranes was expressed as the mean ( SEM) percentage of the total number of cells observed.
Cell survival measurements
The colony assay was used to assess overall cell survival. Single-cell suspensions (0.2 4.0×103 cells per dish, adjusted for irradiation dose) were plated into 60-mm tissue culture dishes, incubated for 3 h, and irradiated to 0 - 10 and 0 - 6 Gy with 60Co -rays (photons) and p(66)/Be neutrons, respectively. After growing for 6 days, colonies were fixed in glacial acetic acid:methanol:water (1:1:8, v/v/v), stained with 0.01% Amido Black in fixative, washed in tap water, air-dried, and were counted. Three independent experiments were performed for each dose point, and the mean surviving fractions were fitted to the linear- quadratic model to generate survival curves. Radiosensitivity was expressed in terms of the mean inactivation dose, D, calculated as the area under survival-dose response curves plotted on a linear-linear scale over the corresponding dose ranges [19]. The mean inactivation dose was chosen over the /β-ratio, as the latter is an indicator of the ‘bendiness’ of the survival curve and often tends to be unrealistically high for neutrons that are not typically low-LET in nature.
mean inactivation dose, D, calculated as the area under survival-dose response curves plotted on a linear-linear scale over the corresponding dose ranges [19]. The mean inactivation dose was chosen over the /β-ratio, as the latter is an indicator of the ‘bendiness’ of the survival curve and often tends to be unrealistically high for neutrons that are not typically low-LET in nature.
Effect of peptide inhibitor P5 on mitochondrial integrity and cell survival
To assess the role of Bak-mediated mitochondrial dissipation in radiosensitivity, matching sets of cultures as described for the colony assay were irradiated in the presence of peptide inhibitor P5 (200 µmol/l) or negative control (200 µmol/l). P5 is a cell-permeable synthetic peptide inhibitor of Bax translocation onto mitochondria, and therefore, was used as a potential suppressor of Bak-mediated mitochondrial pore formation. Both P5 (cat # 1786) and the negative control peptide (cat # 1787) were from Tocris (Bristol, UK). To evaluate the effect of P5 on mitochondrial dissipation, unirradiated controls and samples irradiated to 2.5 Gy of p(66)/Be neutrons or to 6.0 Gy of 60Co -rays were assessed.
The influence of P5 on cell survival was interrogated over the entire range of radiation doses used in the clonogenic assay. The protection against mitochondria-mediated radiation-induced cell death by P5 was expressed as a radioprotection factor (RPF), defined as the ratio

 of mean inactivation doses:
of mean inactivation doses:
RPF = DP5 / D . The subscript P5 indicates irradiation in the
presence of the peptide inhibitor of Bak.
Data analysis
Statistical analyses were performed using the GraphPad Prism software (San Diego, USA) computer program. Standard equations were used to fit nonlinear relationships. To compare two data sets, the unpaired Student’s t-test was used. The p values and the coefficients of determination, R2, were calculated from two-sided tests. A p value of less than 0.05 indicates a statistically significant difference between the data sets.
Results
Radiation-induced mitochondrial dissipation
From the data shown in Figure 1, DU145 cells are moderately susceptible to radiation- induced mitochondrial dissipation. The data show a clear LET-dependent dose-response for mitochondrial dissipation, with a significantly higher proportion of cells with compromised mitochondria per unit dose of neutrons than for the lower LET photons. In both irradiation arms, the extent of mitochondrial dissipation increased sharply with absorbed dose and reached maximum values at 2.5 and 6 Gy for neutrons and photons, respectively (Figure 1). These radiation doses were chosen for further assessment of the impact of peptide inhibitor P5 as they corresponded to the highest achievable radiation-induced mitochondrial dissipation. When cells were irradiated in the presence of peptide inhibitor P5, mitochondrial dissipation was almost completely abolished. The percentage of cells with compromised mitochondria was found to diminish from a maximum of 49 to 17% following both photon (6 Gy) and neutron (2.5 Gy) irradiation. The background level of mitochondrial dissipation in this cell line was found to be high and emerged as 10%.
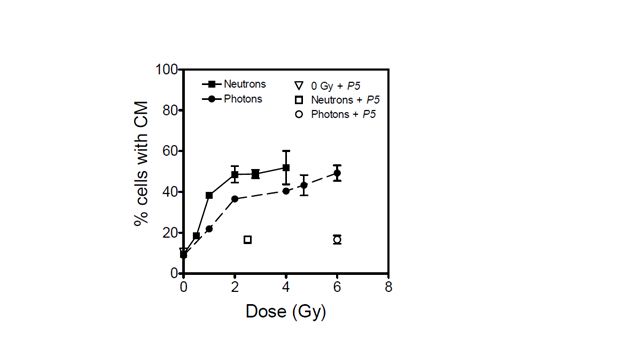
Figure 1: Percentage of DU145 cells with compromised mitochondria (CM) when irradiated with photons (dashed line) and with p(66)/Be neutrons (solid line). The effect of peptide inhibitor P5 on mitochondrial dissipation is shown for 6 Gy of photons (open circle), 2.5 Gy of neutrons (open square), and unirradiated cultures (open triangle). Each data point represents the mean SEM of three independent experiments.
Radioprotection by peptide inhibitor P5
Figure 2 shows cell survival curves for DU145 cell cultures irradiated with or without peptide inhibitor P5. The cell survival parameters are summarised in Table 1. The reduction in radiation-induced mitochondrial dissipation observed in cell cultures irradiated in the presence of P5 (Figure 1), is reflected in an enhancement of radioresistance (Figure 2). The radioprotection factors RPF (based on mean inactivation dose) were found to be 1.360.04 and 1.240.05 for photon and neutron irradiation, respectively, and were not significantly different (P = 0.2222; R2 = 0.2798). On average, cells were rendered 30% more radioresistant by P5, regardless of the type of radiation.
Table 1: Summary of cell survival parameters for human prostate carcinoma cell line, DU145, as obtained from data fitted to the linear-quadratic model. P5 indicates concomitant treatment with a Bak inhibitor. Errors in and β represent the standard error of the best-fit value. Errors in /β-ratios and the radioprotection factor (RPF) were calculated using appropriate error propagation formulae. Values of the mean inactivation dose represent the mean (± SEM) of three independent experiments.
|
Treatment
|
(Gy-1)
|
β (Gy-2)
|
/β (Gy-1)
|
(Gy)
|
RPF
|
|
Photons
|
0.24 ± 0.05
|
0.06 ± 0.01
|
4.00 ± 1.07
|
2.38 ± 0.06
|
1.36 ± 0.04
|
|
Photons + P5
|
0.14 ± 0.06
|
0.05 ± 0.01
|
2.80 ± 1.32
|
3.23 ± 0.04
|
|
Neutrons
|
0.58 ± 0.06
|
0.09 ± 0.01
|
6.44 ± 0.98
|
1.39 ± 0.06
|
1.24 ± 0.05
|
|
Neutrons + P5
|
0.54 ± 0.06
|
0.02 ± 0.01
|
27.00 ± 13.83
|
1.72 ± 0.01
|

Figure 2: Clonogenic survival curves for DU145 cells after photon and p(66)/Be neutron- irradiation in the absence (dashed lines) and presence (solid lines) of peptide inhibitor P5. Each data point represents the mean SEM of three independent experiments.
Discussion
The p53 gene is known to exhibit point mutations and is dysfunctional in DU145 cells [16]. However, wild-type or functional p53 is not always required for cells to undergo mitochondria-mediated apoptosis [6, 7]. In fact, the translocation of Bax or Bak to mitochondria in response to exposure to cytotoxic agents has been demonstrated in cells lacking functional p53 [8, 9]. Bax/Bak translocation leads to dissipation in mitochondrial membrane potential and subsequent apoptosis.
The data presented in Figure 1 show that radiation-induced dissipation of mitochondrial membrane potential increases with radiation dose in a LET-dependent manner. The results also suggest that Bak plays a major role in the compromise of mitochondrial membrane potential in the absence of Bax functionality. Therefore, radiation-induced mitochondrial membrane potential dissipation and subsequent cell death in DU145 cells are likely mediated by translocation of Bak onto mitochondria in a p53-independent manner [8, 9]. Zhang et al. have demonstrated that mitochondrial membrane potential dissipation and cell death after certain stimuli are strictly Bak-dependent [20]. In the absence of p53 functionality, it is also possible that radiation-induced cell death in this cell line occurs via the ceramide pathway for which Bak is activated downstream to regulate apoptosis [21]. These data suggest that Bak-mediated cell death is a major determinant of cellular response to exposure to ionising radiation, and that this phenomenon is independent of the type of radiation (Figures 1 and 2). Inhibition of Bak by P5 resulted in a significant decrease in radiation-induced mitochondrial membrane compromise and a corresponding increase in cell survival.
A factor that plays an important role in the proapoptotic function of Bak is voltage- dependent anion channel 2 (VDAC2). VDAC2 is highly expressed in DU145 cells [22], and is known to inhibit Bak activity [23, 24]. However, in the absence of Bax, as is the case in the cells used in this study, no interaction occurs between VDAC2 and Bak [17]. It is, therefore, not surprising that Bak is central in mediating radiation response in the DU145 cell line, and that its modulation by P5 significantly affects radiation-induced mitochondrial membrane compromise and subsequent cell survival.
The data presented here cannot be corroborated by studies suggesting that high-LET radiation induces p53-independent cell death, with low-LET radiation being more effective in inducing apoptosis in wild-type p53 cells than in mutated p53 and p53-null cells [25, 26]. Neutrons appear to be more potent than 60Co -rays in compromising mitochondrial membrane potential and inducing cytotoxicity (Figures 1 and 2). However, the relative effectiveness of these neutrons is marginal when compared with studies which showed high LET radiation to be ~4 – 6 times more effective than low-LET radiation in inducing apoptosis in cells without p53 functionality [25, 26]. Specifically, the data in Figure 1 show that neutrons induce only 2.22-, 1.42-, and 1.34-fold higher mitochondrial dissipation than 60Co -rays at 1, 2, and 4 Gy, respectively. This modest difference in effectiveness may be due to the relatively large photon-like component of p(66)/Be neutrons [27]. The low LET segment of the lineal energy spectrum (< 10 keV/µm) in p(66)/Be neutrons significantly overlaps with the photon spectrum to the order of 36%, thereby making the neutrons behave more like low-LET radiation. This is consistent with the finding that the radioprotection factor of P5 for photons is only about 10% greater than that for neutrons (Table 1).
Conclusions
In cells that are devoid of p53 and Bax functionality, Bak-mediated mitochondrial dissipation appears to be the main determinant of cellular response to radiation exposure. This is evidenced by the observation that Bak inhibition in p53- and Bax-negative human prostate cancer cells correlates with changes in radiation-induced mitochondrial dissipation, and subsequently with cell survival. Bak inhibition during irradiation almost abolishes mitochondrial dissipation and leads to increased radioresistance. These data seem to indicate that manipulation of Bak availability during irradiation may lead to significant changes in the radiosensitivity. Therefore, targeting Bak availability in tumours might lead to more effective tumour management with radiotherapy.
Acknowledgements
This work was support by grants from the South African National Research Foundation
(grants: No. 85703 and No. 92741).
Conflicts of interest
The authors declare that there are no conflicts of interest associated with this study. The authors alone are responsible for the content and writing of the paper.
References
1. Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene. 2003; 22(37):5897–5906.
2. Choi SY, Kim MJ, Kang CM, Bae S, Cho CK, et al. Activation of Bak and Bax through c-Abl-protein kinase C-p38 MAPK signaling in response to ionizing radiation in human non-small cell lung cancer cells. J Biol Chem. 2006; 281(11):7049–7059.
3. Ubol S, Kramyu J, Masrinoul P, Kachangchaeng C, Pittayanurak P, et al. A novel cycloheptapeptide exerts strong anticancer activity via stimulation of multiple apoptotic pathways in caspase-3 deficient cancer cells. Anticancer Res. 2007; 27(4B):2473–2480.
4. Adrain C, Martin SJ. The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Trends Biochem Sci. 2001; 26(6):390–397.
5. Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005; 121(5):671-674.
6. Han Z, Chatterjee D, He DM, Early J, Pantazis P, et al. Evidence for a G2 checkpoint in p53-independent apoptosis induction by X-irradiation. Mol Cell Biol. 1995; 15(11):5849–5857.
7. Chapman JR, Tazaki H, Mallough C, Konno S. Brefeldin A-induced apoptosis in prostatic cancer DU-145 cells: a possible p53-independent death pathway. BJU Int. 1999; 83(6):703–708.
8. Strobel T, Swanson L, Korsmeyer S, Cannistra SA. BAX enhances paclitaxel- induced apoptosis through a p53-independent pathway. Proc Natl Acad Sci USA. 1996; 93(24):14094–14099.
9. Lee YJ, Chung DY, Lee SJ, Jhon GJ, Lee, YS. Enhanced radiosensitization of p53 mutant cells by oleamide. Int J Radiat Oncol Biol Phys. 2006; 64(5):1466–1474.
10. Harima Y, Harima K, Shikata N, Oka A, Ohnishi T, et al. Bax and Bcl-2 expressions predict response to radiotherapy in human cervical cancer. J Cancer Res Clin Oncol. 1998; 124(9):503–510.
11. Mackey TJ, Borkowski A, Amin P, Jacobs SC, Kyprianou, N. Bcl-2/bax ratio as a predictive marker for therapeutic response to radiotherapy in patients with prostate cancer. Urology. 1998; 52(6):1985–1090.
12. Wootipoom V, Lekhyananda N, Phungrassami T, Boonyaphiphat P, Thongsuksai P. Prognostic significance of Bax, Bcl-2, and p53 expressions in cervical squamous cellcarcinoma treated by radiotherapy. Gynecol. Oncol. 2004; 94(3):636–642.
13. Khor LY, Moughan J, Al-Saleem T, Hammond EH, Venkatesan, V, et al. Bcl-2 and Bax expression predict prostate cancer outcome in men treated with androgen deprivation and radiotherapy on Radiation Therapy Oncology Group Protocol 92-02. Clin Cancer Res. 2007; 13(12):3585–3590.
14. Khor LY, DeSilvio M, Li R, McDonnell TJ, Hammond EH, et al. Bcl-2 and Bax expression and prostate cancer outcome in men treated with radiotherapy in Radiation Therapy Oncology Group Protocol 86-10. Int J Radiat Oncol Biol Phys. 2006; 66(1):25–30.
15. Neise D, Graupner V, Gillissen BF, Daniel PT, Schulze-Osthoff K. Activation of the mitochondrial death pathway is commonly mediated by the preferential engagement of Bak. Oncogene. 2008; 27(10):1387–1396.
16. Carroll AG, Voeller HJ, Sugars L, Gelmann E. p53 oncogene mutations in three human prostate cell lines. The Prostate. 1993; 23(2):123–134.
17. Chandra D, Choy G, Daniel PT, Tang DG. Bax-dependent regulation of bak by voltage-dependent anion channel 2. J Biol Chem. 2005; 280(19):19051–19061.
18. Hemmati PG, Güner D, Gillissen B, Wendt J, von Haefen C, et al. Bak functionally complements for loss of Bax during p14ARF-induced mitochondrial apoptosis in human cancer cells. Oncogene. 2006; 25(50):6582–6594.
19. Akudugu JM, Slabbert JP, Roos WP, Böhm L. Micronucleus response of human glioblastoma and neuroblastoma cells toward low-LET photon and high-LET p(66)/Be neutron irradiation. Am J Clin Oncol Canc. 2003; 26(3):e1–e6.
20. Zhang W, Wang X, Chen T. Resveratrol induces apoptosis via a Bak-mediated intrinsic pathway in human lung adenocarcinoma cells. Cell Signal. 2012; 24(5):1037–1046.
21. Husari AW, Dbaibo GS, Bitar H, Khayat A, Panjarian S, et al. Apoptosis and the activity of ceramide, Bax and Bcl-2 in the lungs of neonatal rats exposed to limited and prolonged hyperoxia. Resp Res. 2006; 7:100.
22. Liu Z, Marquez M, Nilsson S, Holmberg AR. Comparison of protein expression in two prostate cancer cell-lines, LNCaP and DU145, after treatment with somatostatin. Oncol Rep. 2009; 22(6):1451–1458.
23. Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003; 301(5632):513–517.
24. Ren D, Kim H, Tu HC, Westergard TD, Fisher JK, et al. The VDAC2-BAK rheostat controls thymocyte survival. Sci Signal. 2009; 2(85):ra48.
25. Takahashi A, Matsumoto H, Furusawa Y, Ohnishi K, Ishioka N, et al. Apoptosis induced by high-LET radiations is not affected by cellular p53 gene status. Int J Radiat Biol. 2005; 81(8):581–586.
26. Takahashi A, Matsumoto H, Yuki K, Yasumoto JI, Kajiwara A, et al. High-LET radiation enhanced apoptosis but not necrosis regardless of p53 status. Int J Radiat Oncol Biol Phys. 2004; 60(2):591–597.
27. Slabbert JP, Theron T, Serafin A, Jones DT, Böhm, L, et al. Radiosensitivity variations in human tumour cell lines exposed in vitro to p(66)/Be neutrons or Co--rays. Strahlenther Onkol. 1996; 172(10):567–572.