Review
2015
December
Volume : 3
Issue : 4
Clinical and genetic heterogeneity in Wilson disease - A review
Advithi Rangaraju, Sridhar MS, Poonam Naik
Pdf Page Numbers :- 192-197
Advithi Rangaraju1,*, Sridhar MS2 and Poonam Naik1
1KIMS Foundation and Research Centre, Minister Road, Secunderabad-500003, Telangana, India
2Department of Ophthalmology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India
*Corresponding author: Dr. Advithi Rangaraju, Ph.D., Scientist (Genetics), KIMS Foundation and Research Centre, Minister Road, Secunderabad-500003, Telangana, India. Email: advithi.rangaraju@gmail.com
Received 22 June 2015; Revised 19 August 2015; Accepted 27 August 2015; Published 5 September 2015
Citation: Advithi Rangaraju, Sridhar MS, Poonam Naik. Clinical and genetic heterogeneity in Wilson disease - A review. J Med Sci Res. 2015; 3(4):192-197. DOI: http://dx.doi.org/10.17727/JMSR.2015/3-037
Copyright: © 2015 Advithi Rangaraju, et al. Published by KIMS Foundation and Research Center. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Wilson disease (WD) is an inborn error of copper metabolism leading to its accumulation in liver, kidney and cornea. It is caused by a defective ATPase protein which is coded by ATP7B gene. It follows an autosomal recessive mode of inheritance with a prevalence of 1 in 30,000. WD shows varied clinical heterogeneity making clinical diagnosis a difficult task. The corneal Kayser-Fleischer (KF) ring is an important diagnostic criterion as it is invariably present in 95% of the WD cases. In this review, we discussed the varied clinical manifestations of WD which makes diagnosis a challenging process. Though genetic testing is a reliable technique to confirm clinical diagnosis, genotype-phenotype correlations are yet to be established. This could be attributed to the consanguinity and ethnic variation observed in the Indian population, suggesting genetic heterogeneity leading to clinical heterogeneity making diagnosis difficult. Further, genetic studies are warranted to establish genotype-phenotype correlations which can pave way for early diagnosis and treatment. Genetic testing will help in identifying pre-symptomatic siblings and other family members of the patient who should be advised for regular follow-up. A combination of clinical and genetic studies should be considered for proper understanding of disease manifestation and for making an early clinical diagnosis of WD.
Keywords: Wilson disease; copper metabolism; genetic testing; ATP7B gene; KF ring
Full Text
Background
Copper is an essential trace element in prokaryotic and eukaryotic cells, where over thirty proteins, including the electron transport enzymes, are known to require its specific oxidative capacity to perform their normal biological roles. When copper homeostasis is disrupted, this same oxidative potential can induce the production of highly reactive free radicals with concomitant extensive cellular damage [1].
Wilson disease is a rare autosomal recessive disorder of copper metabolism, with a prevalence of about 1 in 30,000 people. It is characterized by a decreased biliary copper excretion and a defective incorporation of copper into ceruloplasmin, leading to copper accumulation in the liver, brain and kidneys [2]. WD is present in most populations worldwide, and particularly in those in which consanguineous marriage is common. The disease frequency is estimated to be between 1 in 30,000, and the carrier frequency is approximately 1 in 90.
Clinical presentation is highly heterogeneous and show predominantly hepatic, neurologic, psychiatric and ophthalmological manifestations, with many patients having a combination of symptoms [3, 4]. It typically begins with a pre-symptomatic period, during which copper accumulation in the liver causes subclinical hepatitis, and progresses to liver cirrhosis and development of neuropsychiatric symptoms. It may also present as fulminant hepatic failure with an associated Coombs-negative haemolytic anaemia and acute renal failure. Many unique case reports on Wilson disease with respect to the clinical diagnosis and manifestations have been published so far. Likewise, genetics of WD has also been widely studied with more than 500 mutations reported in the Wilson disease database so far. In this review, we will discuss the varied clinical and genetic manifestations and what may determine the underlying heterogeneity.
Clinical manifestations
Hepatic
Hepatic dysfunction in Wilson disease may assume several forms. Asymptomatic enlargement of the liver and spleen may occur, sometimes with elevation of liver enzymes. Acute transient hepatitis is the mode of presentation in 25% of those in whom hepatic symptoms herald disease onset. The hepatic symptoms of Wilson’s disease may also mimic autoimmune hepatitis [5, 6]. Hemolysis, is also a common finding in patients with acute liver failure due to Wilson disease, but sometimes may occur episodically independent of liver failure [7].
Neurological
A variety of psychiatric symptoms may appear in the setting of Wilson’s disease. Personality changes and disturbances of mood, particularly depression, are the most frequent behavioral features of Wilson’s disease [8]. Tremor, which may be resting, postural, or kinetic, is the most common initial neurological feature of Wilson’s disease. Dysarthria is also common in persons with Wilson’s disease and may possess either an extra pyramidal or a cerebellar character [9, 10]. A variety of other neurological features may emerge in Wilson’s disease. Cerebellar dysfunction develops in 25% of individuals with neurological Wilson’s disease. Dystonia may be present in almost 40%. Gait abnormalities are a frequent component of neurological Wilson’s disease [11].
Ophthalmic
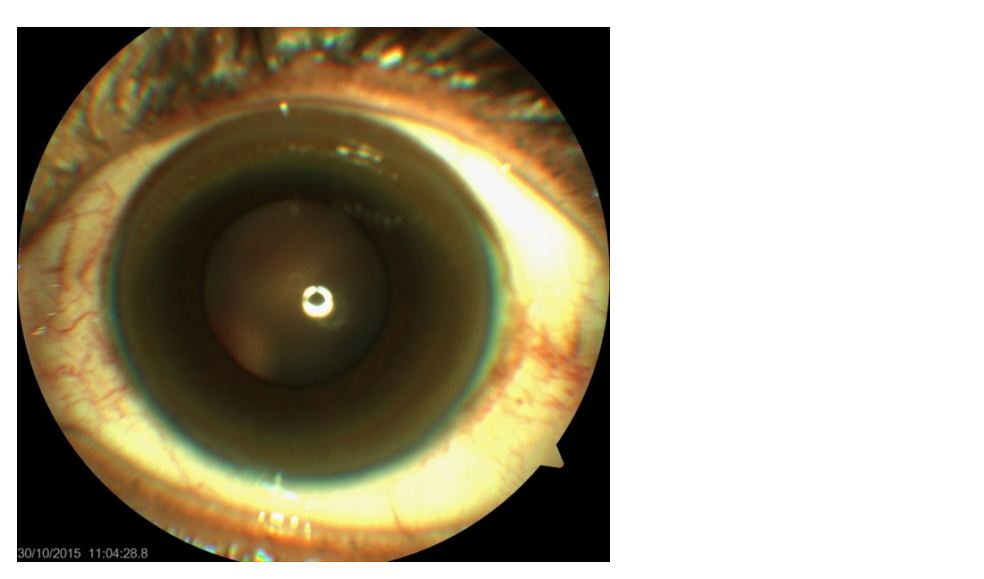
The ophthalmological manifestation of Wilson’s disease is Kayser-Fleischer rings. Kayser-Fleischer rings are formed by deposition of copper within Descemet’s membrane of the cornea (Figure 1). Excess copper is actually deposited throughout the cornea in Wilson’s disease, but it is only in Descemet’s membrane that sulfur-copper complexes are formed, producing the visible copper deposits. The other classic ophthalmological manifestation of Wilson’s disease is the sunflower cataract, which was first described by Siemerling and Oloff in 1922, wherein, copper deposition in the lens assumes a sunburst or sunflower appearance, with a central disc and radiating petal-like spokes [12].
Radiological manifestations
Wilson disease also has bone and joint involvement, wherein radiographic evidence of osteoporosis is present in up to 88% of persons with Wilson’s disease resulting in spontaneous fractures [13]. Joint involvement, especially at the knees, is also common and joint pain may be the presenting symptom of Wilson’s disease [14]. Radiological evidence of vertebral column abnormalities is evident in 20 to 33% of individuals with Wilson’s disease [15]. The most common radiological abnormality was a generalized increase of radiolucency, interpreted as skeletal demineralization followed by premature osteoarthrosis. Changes in the spine were common and included osteochondritis, reduction of intervertebral joint spaces, osteoarthrosis, and a tendency to squaring of vertebral bodies. Other bony changes included fluffy irregularity of femoral trochanters, osteochondritis dissecans of the knees, osteophytic protrusions at bone ends, and bunch of tongue-like osteophytes at joint margins. The symptoms associated with these radiological abnormalities comprised back pain and stiffness with restricted movement, pain and stiffness of knees, hips, and wrists, and tenderness to pressure over margins of affected joints. Joint hypermobility was also observed and episodes of acute polyarthritis with serological changes were also seen [13].
Other manifestations
Haemolytic anaemia, presumably due to copper induced oxidative damage to erythrocytes, may be the initial manifestation of Wilson’s disease in 10 to 15% of cases [16]. Thrombocytopenia may also develop, either separately or in conjunction with haemolytic anaemia [17]. A patient with thrombocytopenia and the combination of Wilson’s disease and anti-phospholipid antibody syndrome has also been reported [18]. Renal involvement may also occur in Wilson’s disease manifesting as renal tubular dysfunction, with consequent hypercalciuria and hyperphosphaturia, which may induce nephrocalcinosis [19]. Hypokalemia with muscle weakness and even respiratory failure has also been reported in Wilson’s disease, presumably secondary to renal tubular dysfunction [20]. A skin change with hyper pigmentation of the anterior lower legs, potentially misinterpreted as Addison’s disease, has been reported in a patient with Wilson’s disease [21]. Some rare manifestations like gynecological abnormalities (menstrual irregularity, delayed puberty, gynecomastia, cardiovascular dysfunction (congestive heart failure, cardiac arrhythmia), and other impairments such as glucose intolerance, parathyroid are also reported [22, 23]. Even within a given family, patients often present with different symptoms. There is wide variability in the reported rates of the different clinical manifestations seen at the time of presentation.
Etiology
Wilson’s disease is caused by a mutation in the ATP7B gene that code for a membrane-bound, P-type copper-transporting ATPase expressed primarily in the liver. The mutations cause a decrease or absence of the gene product or production of a protein with impaired function. This results in reduced copper transport within the hepatocyte and failed insertion of copper into ceruloplasmin. As a consequence, in most patients with Wilson’s disease serum ceruloplasmin and total serum copper concentrations are low, but there is an increased proportion of low molecular weight copper in the serum. This eventually allows deposition of excess copper in the liver, brain, eye, and other tissues, leading to the variable clinical symptoms. Absence of the protein in the kidney impairs tubular re-absorption of filtered copper with resultant increased urine copper excretion. Excess copper damages mitochondria, which produces oxidative damage to cells and allows spillage of copper into the blood, thereby overloading other organs such as the brain, kidney and red blood cells, initiating toxic damage [4].
In WD, apoptotic cell death is also accelerated by the inhibition of IAPs (inhibitor of apoptosis proteins) that is caused by toxic deposits of intracellular copper [24]. Normally, IAPs inhibit caspase-3 and caspase-7, which are responsible for apoptotic cell death. The main areas of the brain affected in WD are the lenticular nuclei, which macroscopically appear brown in color because of copper deposition. Degeneration occurs with disease progression, leading to necrosis, gliosis and cystic changes, and lesions can be seen in the brainstem, thalamus, cerebellum and cerebral cortex. With disease progression, copper deposits lead to vacuolar degeneration in proximal renal tubular cells, causing ‘Fanconi syndrome’ and the appearance of the golden-brown ‘Kayser–Fleischer’ (KF) ring in Descemet’s membrane in the cornea. Acute release of copper into the circulation can damage red blood cells, thereby inducing hemolysis.
Diagnosis
Clinical symptoms (hepatic symptoms, neurologic symptoms, and cornea Kayser-Fleischer ring) and biochemical tests (elevated 24-h urinary copper, low plasma ceruloplasmin, and elevated liver copper concentrations) are done for the diagnosis of WD. However, liver biopsy is considered to be the gold standard for the diagnosis of WD. Biochemical markers can be sometimes misleading rendering WD diagnosis difficult in the absence of typical symptoms. Genetic testing may be helpful to establish a difficult diagnosis [25-27].
Histopathology
The earliest changes detectable with light microscopy include glycogen deposition in the nuclei of periportal hepatocytes and moderate fatty infiltration. The lipid droplets, which are composed of triglycerides, progressively increase in number and size, sometimes resemble the steatosis induced by ethanol. Hepatocyte mitochondria typically exhibit heterogeneity in size and shape, with increased matrix density, separation of the normally apposed inner and outer mitochondrial membranes, widened intercristal spaces, and an array of vacuolated and crystalline inclusions within the matrix. With progression of disease, copper protein is sequestered in lysosomes and is visible as electron-dense pericanalicular granules. Hepatocytes at the periphery of the nodules frequently contain Mallory hyalin. One proposed mechanism implicates copper as the inducer of fibrogenesis.
In neurological case gross anatomical changes has been observed include degeneration and cavitation, primarily involving the putamen, globus pallidus, caudate nucleus, and thalamus. Little correlation has been observed between the degree of neurologic impairment and the neuropathologic findings. The affected areas of the brain do not possess higher copper concentrations than the unaffected portions.
Kayser-Fleischer rings result from copper deposition in Descemet's membrane of the cornea and reflect a high degree of copper storage in the body and are the most unique sign of Wilson disease. During an eye exam, an ophthalmologist will see a rusty-brown ring around the edge of the iris and in the rim of the cornea. The iris is the colored part of the eye surrounding the pupil. The cornea is the transparent outer membrane or layer that covers the eye. People with Wilson disease who show signs of nervous system damage usually have Kayser-Fleischer rings.
Electron microscopy
Electron microscopic studies on ultrathin sections of liver reveal numerous electron-dense lysosomes and residual bodies. The abnormality found in all cells appeared mainly in the pericanalicular zone where dark bodies were observed [28]. The elemental analysis in transmission electron microscopy with electron energy loss spectroscopy, and in scanning electron microscopy with energy dispersive x-ray analysis, shows copper-specific signals of electron-dense accumulations inside these dark lysosomes and residual bodies.
The electron microscopic detection of copper-containing hepatocytic lysosomes is helpful in the diagnosis of the early stages of Wilson disease, in addition to the quantification of hepatic copper by atomic absorption spectrophotometry. In case of eye the cornea from one eye of the patients is divided into five anatomic regions--superior, inferior, medial, lateral, and central--and each area is examined by x-ray energy spectroscopy interfaced with transmission electron microscopy and by atomic absorption spectrophotometry. Electron-dense granules, rich in copper and sulfur, is present in both the peripheral and the central region of the cornea but were more numerous at the periphery [29].
Genetics
Mutations in ATP7B gene have been implicated in Wilson disease. It is a relatively large gene of 80 kb, comprised of 21 exons and consists of six metal-binding domains, eight transmembrane segments, an ATP-binding domain typical of copper ATPases with a P-domain an N-domain, and an A-domain with the TGE sequence motif [30]. It is responsible for transporting copper from intracellular chaperon protein into the secretory pathway for excretion into bile and incorporation into apo-ceruloplasmin for the synthesis of functional ceruloplasmin [31].
A variety of defects have been identified in the ATP7B gene of WD patients, the majority of which are located in the transmembrane region of the associated protein. These defects include insertion, deletion, splice site and point mutations. Many of the mutations identified are described in various databases. It has been noted that genetic heterogeneity in patients with WD exists across various races and geographical regions. In most ethnic groups, either one or a small number of these ATP7B mutations are prevalent, in addition to many other more rare mutations. For instance, among Europeans and North Americans, two ATP7B point mutations— His1069→Gln and Gly1267→Arg—together account for 38% of the observed mutations in WD [32]. A 15-nucleotide deletion underlying the most common haplotype seen in Sardinians has been reported, and has a missense mutation (Arg778→Leu) found among Mongoloids [33]. Interestingly, none of these two mutations have been found in Indians.
Seven recurring haplotypes were identified accounting for 58% of the different mutant chromosomes in WD in Indians [34]. Till date, a total of 51 mutations have been documented in India including 34 novel mutations (Chandigarh-18, Kolkata-5, and Vellore-11). Of which C813A is the most common mutation. There is no single predominant mutation noted in the Indian population unlike the studies in other countries suggesting genetic heterogeneity. Exons 2, 8, 13, 14, and 18 are reported to be hotspots from Indian studies which accounted for the majority of mutations (86.4%). Exons 13 and 18 were considered to be the hotspots for the South Indian population [34, 35]. However, lack of dominant mutations prevented the genotype-phenotype correlation in this study [35].
WD is perhaps more common than reported from India, therefore, epidemiological and multicentre studies are needed in view of high degree of consanguinity found particularly in south India.
Conclusion
Wilson disease is an autosomal recessive genetic disorder with varied clinical and genetic manifestations. Most of the patients are diagnosed at the end stage of liver cirrhosis, wherein liver transplant becomes the only option which adds to a huge economic burden apart from the high mortality rates. Diagnosing WD in the early stages is the best option possible with consistent patient follow-up. However, despite molecular genetics being a reliable technique for confirmation of the WD diagnosis, genotype-phenotype correlation in pre-symptomatic siblings is still a challenge due to the absence of similar dominant mutations in the siblings. Further, family studies are warranted to better understand the genotype-phenotype correlation in patients with WD. A national mutation database may improve the awareness and lead to early diagnosis and prompt treatment. Genetic testing should be recommended in any unexplained liver and neurological manifestations which should be followed by family screening. This should be followed by genetic counseling and regular follow-up in the pre-symptomatic siblings, which is the appropriate way for early clinical diagnosis.
Acknowledgements
The contribution of all the authors is acknowledged in preparation of the manuscript.
Conflict of interest
Authors declare no conflict of interest.
References
1. Sarkar B. Transport of copper. In Sigel H. (ed.), Metal ions in biological systems. Marcel Dekker, New York. 1981; 233–281.
2. Scheinberg IH, Sternlieb I. Wilson’s disease. In: Smith Jr LH, editor. Major problems in internal medicine, vol. 23. Philadelphia, PA: WB Saunders, 1984; p. 25–35.
3. Riordan SM, Williams R. The Wilson's disease gene and phenotypic diversity. J Hepatol. 2001; 34:165–171
4. Schilsky M, Tavill AS. Wilson disease. In Disease of the Liver, edn 9, (Eds Schiff ER et al.) Philadelphia: Lippincott Williams & Wilkins, 2003; p. 1169–1186.
5. Sternlieb I, Scheinberg IH. Chronic hepatitis as a first manifestation of Wilson’s disease. Ann Intern Med. 1972; 76(1):59–64.
6. Scott J, Gollan JL, Samourian S, Sherlock S. Wilson’s disease presenting as chronic active hepatitis. Gastroenterology. 1978; 74(4):645–651.
7. Takeshita Y, Shimizu N, Yamaguchi Y, Nakazono H, Saitou M, et al. Two families with Wilson disease in which siblings showed different phenotypes. J Hum Genet. 2002; 47(10):543–547.
8. Brewer GJ. Behavioral abnormalities in Wilson’s disease. Adv Neurol. 2005; 96:262–274.
9. Walshe JM, Yealland M. Wilson’s disease: the problem of delayed diagnosis. J Neurol Neurosurg Psychiatry. 1992; 55(8):692–696.
10. Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002; 22:439–458.
11. Svetel M, Kozic D, Stefanova E, Semnic R, Dragasevic N, et al. Dystonia in Wilson’s disease. Mov Disord. 2001; 16(4):719–723.
12. Wiebers DO, Hollenhorst RW, Goldstein NP. The ophthalmologic manifestations of Wilson’s disease. Mayo Clin Proc. 1977; 52(7):409–416.
13. Golding DN, Walshe JM. Arthropathy of Wilson’s disease: study of clinical and radiological features in 32 patients. Ann Rheum Dis. 1977; 36(2):99–111.
14. Canelas HM, Carvalho N, Scaff M, Vitule A, Barbosa ER, et al. Osteoarthropathy of hepatolenticular degeneration. Acta Neurol Scand. 1978; 57(6):481–487.
15. Mindelzun R, Elkin M, Scheinberg IH, Sternlieb I. Skeletal changes in Wilson’s disease: a radiological study. Radiology. 1970; 94(1):127–132.
16. Yarze JC, Martin P, Munoz SJ, Friedman LS. Wilson’s disease: current status. Am J Med 1992; 92(6):643–654.
17. Donfrid M, Jankovic G, Strahinja R, Colovic R, Begic- Janeva A, et al. Idiopathic thrombocytopenia associated with Wilson’s disease. Hepatogastroenterology. 1998; 45(23):1774–1776.
18. Atanassova PA, Panchovska MS, Tzvetanov P, ChalakovaNT, Masaldzhieva RI, et al. Hepatolenticular degeneration combined with primary antiphospholipid syndrome: a case report. Eur Neurol. 2006; 55(1):42–43.
19. Wiebers DO, Wilson DM, McLeod RA, Goldstein NP. Renal stones in Wilson’s disease. Am J Med. 1979; 67(2):249– 254.
20. Narayanan P, Chetan G, Mahadevan S. Wilson’s disease presenting as respiratory failure. Indian J Pediatr 2006; 73(1):99–100.
21. Leu ML, Strickland GT, Wang CC, Chen TS. Skin pigmentation in Wilson’s disease. JAMA 1970; 211(9):1542–1543.
22. Scheinberg IH, Sternlieb I. Wilson’s disease. Annu Rev Med. 1965; 16:119–134.
23. Lau JY, Lai CL, Wu PC, Pan HY, Lin HJ, et al. Wilson’s disease: 35 years’ experience. Q J Med. 1990; 75(278):597–605.
24. Mufti AR, Burstein E, Csomos RA, Graf PC, Wilkinson JC, et al. XIAP is a copper binding protein deregulated in Wilson’s disease and other copper toxicosis disorders. Mol Cell. 2006; 21(6):775–785.
25. Roberts, Eve A, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology, 2008; 47(6): 2089–2111.
26. Medici V, Rossaro L, Sturniolo GC. Wilson disease—a practical approach to diagnosis, treatment and follow-up. Digestive and liver disease. 2007; 39(7): 601–609.
27. Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. The Lancet. 2007; 369(9559):397–408.
28. Schaffner F, Sternlieb, Barka T, Popper H. Hepatocellular changes in Wilson's disease. Am. J. Path. 1962; 41:315.
29. Johnson RE, Campbell RJ. Wilson's disease. Electron microscopic, x-ray energy spectroscopic, and atomic absorption spectroscopic studies of corneal copper deposition and distribution. Lab Invest. 1982; 46(6):564–569.
30. Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007; 87(3):1011–1046.
31. Tao TY, Gitlin JD. Hepatic copper metabolism: insights from genetic disease. Hepatology. 2003; 37(6):1241–1247.
32. Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995; 9(2):210–217.
33. Kim MS, Repp A, Smith DP. LUSH odorant-binding protein mediates chemosensory responses to alcohols in Drosophila melanogaster. Genetics; 1998; 150(2):711–721.
34. Gupta A, Aikath D, Neogi R, Datta S, Basu K, et al. Molecular pathogenesis of Wilson Disease: haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Hum Genet. 2005; 118(1):49–57.
35. Santhosh S, Shaji RV, Eapen CE, Jayanthi V, Malathi S, et al. ATP7B mutations in families in a predominantly Southern Indian cohort of Wilson's disease patients. Indian J Gastroenterol. 2006; 25(6): 277–282.