Full Text
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive and life-threatening syndrome of excessive immune activation. It most frequently affects infants from birth to 18 months of age, but the disease is also observed in children and adults of all ages. HLH can occur as a familial or sporadic disorder and it can be triggered by a variety of events that disrupt immune homeostasis. Infection is a common trigger both in those with a genetic predisposition and in sporadic cases [1].
Case report
62-years-old female, presented with shortness of breath, cough, fever for one month duration, history of weight loss (5 kg) & loss of appetite, no hemoptysis, no chest pain, no abdominal pain, no malena, no genitourinary symptoms. History of Left pleural effusion- tubercular one year back, taken anti-tubercular treatment (ATT) for 3 months then discontinued. Known case of diabetes mellitus, hypothyroid. On admission patient was tachypneic, pallor, bilateral pedal edema, RS– bilateral crepitations, P/A- Hepatomegaly were present. Oxygen saturation low (Spo2–80% at room air), hypotensive, initiated on noninvasive ventilation (NIV) and inotrope support. Blood workup revealed anemia (8.6 gm%), leucopenia (1900 cells/mm3), platelets count –1.62 lakhs, elevated serum creatinine (2.65mg/dl), elevated liver enzymes (SGPT- 121, SGOT-69), low albumin level (1.8gm/dl), serum procalcitonin- 27.61, prothrombin time -14.1sec, inr -1.23, abg – s/o metabolic acidosis. Chest X-ray showed B/L mid and lower zone dense opacities (Figure 1).
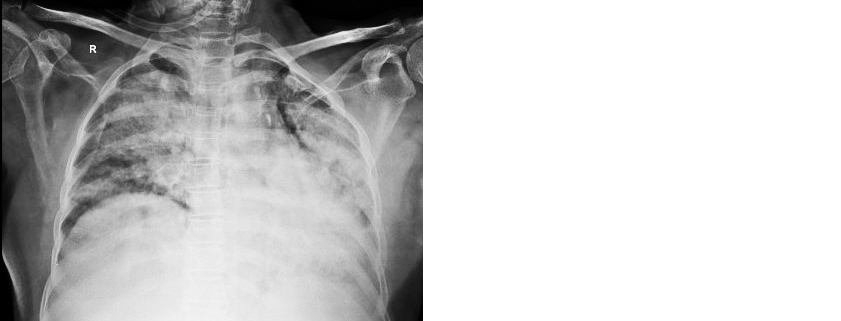
Figure 1: Chest radiograph showing bilateral consolidation.
Started on I.V antibiotics (pipercillin + tazobactum), nebulized bronchodilators. She did not tolerate non-invasive ventilation (NIV), so intubated and connected to mechanical ventilatory support. 2D Echo - normal. Serological workup for HIV, Hep B & C negative. Bronchoscopy done, bronchial wash CBNAAT MTB detected, and no rifampicin resistance. So started on ATT accordingly in renal doses. Bronchial wash for pyogenic culture - no growth. Ultrasound abdomen showed hepatosplenomegaly, ascites. Serial blood investigation revealed thrombocytopenia, elevated serum ferritin levels (1989 ng/ml), hypertriglyceridemia (597 mg/dl), serum fibrinogen – 232 mg/dl, prolonged prothrombin time (20.6 sec) and INR- 1.79. In view of worsening cytopenia and hepatosplenomegaly, bone marrow biopsy done – reports s/o granulomatous inflammation with associated haemophagocytic syndrome (Figures 2 and 3). She deteriorated, neurological status worsened, unfortunately patient expired on 5th day of admission.
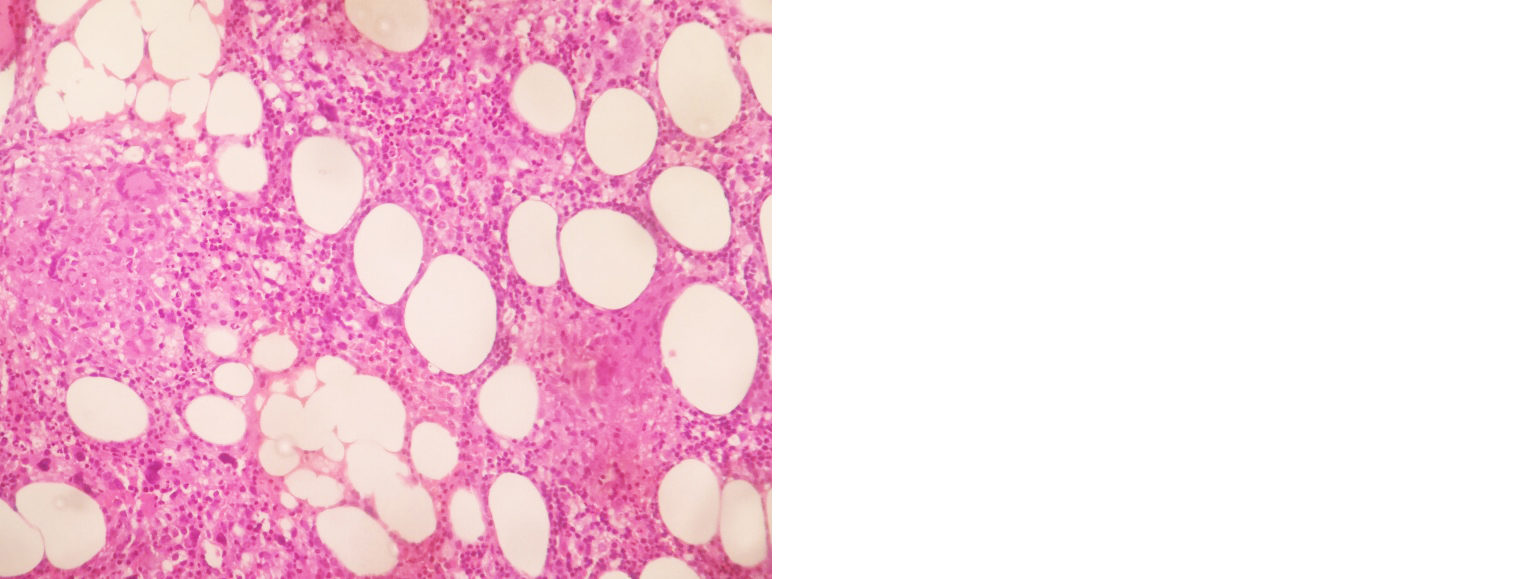
Figure 2: Bone marrow biopsy slide showing epithelioid cell granulomas.
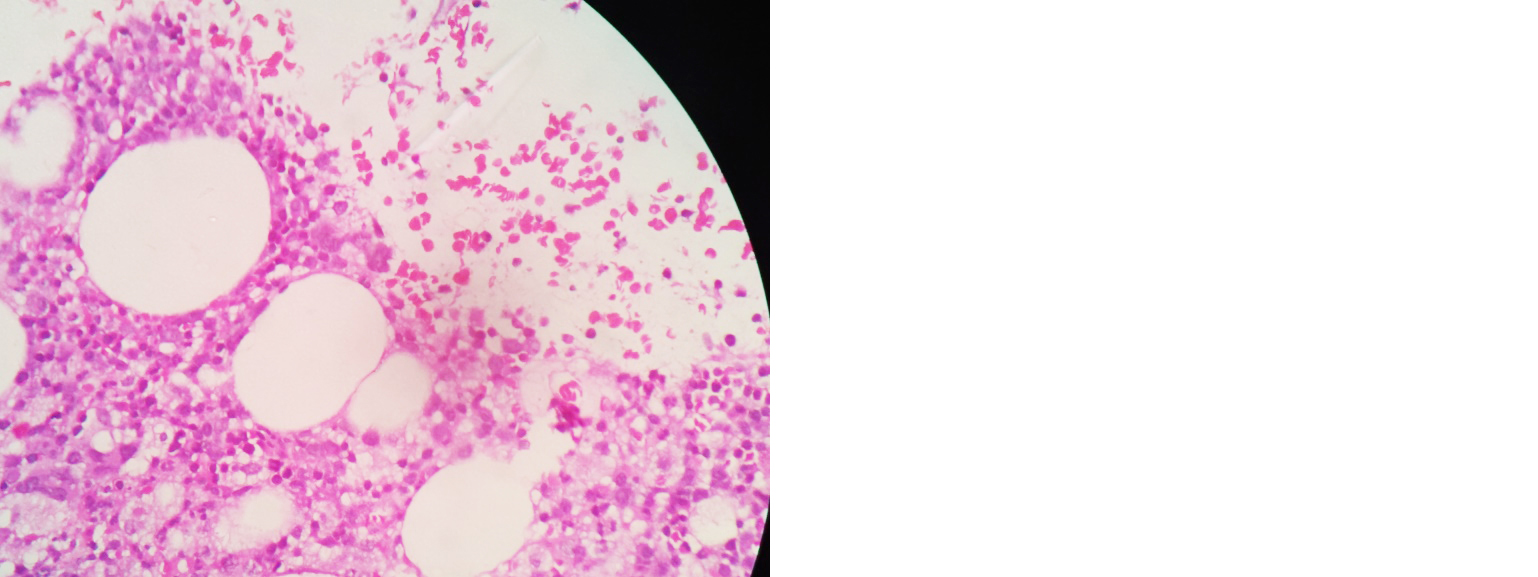
Figure 3: Bone marrow biopsy slide showing plenty of activated macrophages with focal phagocytosis.
Discussion
Hemophagocytic lymphohistiocytosis (HLH) is a rare, aggressive, life threatening syndrome of excessive inflammation and tissue destruction due to abnormal immune activation. There are two types: primary (Genetic) HLH – due to known and unknown genetic defects (Familial HLH) and immune deficiency syndromes (Chediak Higashi syndrome, Griscelli syndrome, X linked Immunoproliferative syndromes). Secondary HLH(acquired) describes patient with HLH phenotype in absence of known genetic cause, typically occurs in setting of infections, autoimmune & malignant diseases, immunosuppression/ organ transplantation [2, 3].
Normally natural killer cells (constitutes 10–15% of lymphocytes) and cytotoxic lymphocytes (activated T lymphocytes) regulates and eliminate damaged, infected host cells such as macrophages bearing foreign antigen. In HLH these two fails to eliminate activated macrophages, this lack of normal feedback regulation leads to excess macrophage activity & elevated levels of cytokines [4, 5], resulting in tissue damage. Haemophagocytosis is characterized by presence of RBC, platelets or WBC or fragments of these cells within cytoplasm of macrophages. Elevated cytokines include INF gamma, TNF alpha, interleukins such as IL-6, IL-10, IL-12 and soluble IL-2 receptor (CD25). HLH presents as febrile illness associated with multiple organ involvement. Primary HLH restricted to young age (80% presents in <1 year), in adults almost all cases are secondary. Diagnostic criteria [6] includes five of following 8 findings: 1. Fever (≥38.5o C), 2. Splenomegaly, 3. Peripheral blood cytopenia (at least two of following – Hb <9g/dl; platelets<100,000/µL; Absolute neutrophil count <1000/µL), 4. Hypertriglyceridemia (Fasting TG >265mg/dl) and/or hypofibrinogenemia (Fibrinogen <150mg/dl), 5. Hemophagocytosis in bone marrow, spleen, lymph node or liver, 6. Low or absent NK cell activity, 7. Elevated serum ferritin (>500ng/mL), 8. Elevated soluble CD25 (>2400U/mL) or molecular identification of an HLH associated gene mutation eg., PRF mutations, SAP mutations. Additional findings that are common are transaminitis, coagulopathy, hyponatremia, edema, rash, hypoalbuminemia, elevated lactate dehydrogenase(LDH), C-reactive protein, decreased high density lipoprotein, elevated cerebrospinal fluid protein and cells and neurologic symptoms ranging from focal deficits to altered mental status [7, 8]. Treatment includes suppressing the severe inflammation (Immunosuppressive agents – dexamethasone, cyclosporine, etoposide) and treating the triggering agent (infection- antibiotics/ antiviral drugs, neoplasm). Patients with genetic mutations/ familial HLH allogenic hematopoietic stem cell transplantation can be done.
Disseminated tuberculosis (TB) is defined as having two or more noncontiguous sites resulting from lymphohematogenous dissemination of Mycobacterium tuberculosis, clinical manifestations are most likely of subacute or chronic, less commonly acute presentation may present. Acute miliary TB may be fulminant including multiorgan system failure and acute respiratory distress syndrome (ARDS) [9, 10].
In our case, patient had disseminated kochs, sepsis, so started on antibacterial and anti-tubercular therapy. Cytopenias, hepatosplenomegaly raised the suspicion of HLH, so further evaluated. 6 out of 8 findings in diagnostic criteria for HLH were positive, diagnosed to have secondary HLH due to disseminated tuberculosis.
Conclusion
Though in our case - tuberculosis sepsis, a rare complication of disseminated tuberculosis is the differential diagnosis, presence of classical features such as hemophagocytosis in bone marrow, cytopenias, splenomegaly, hyperferritinemia, hypertriglyceredemia strongly suggest hemophagocytic lymphohistiocytosis. Making the correct diagnosis is crucial, because treatment differs from sepsis, as HLH needs immunosuppressive therapy. Prompt initiation of immunochemotherapy is essential but timely diagnosis may be challenging because of rarity of HLH.
Acknowledgement
The Department of Radiology & Imageology, Krishna Institute of Medical Sciences (KIMS), Secunderabad-500003, Telangana, India. Conflicts of interest Authors declare no conflicts of interest.
References
[1] Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant 2010; 16(1 Suppl):S82–S89.
[2] Rivière S, Galicier L, Coppo P, Marzac C, Aumont C, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med 2014; 127(11):1118–1125.
[3] Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet 2014; 383(9927):1503–1516.
[4] Pachlopnik Schmid J, Côte M, Ménager MM, Burgess A, Nehme N, et al. Inherited defects in lymphocyte cytotoxic activity. Immunol Rev 2010; 235(1):10–23.
[5] Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr 2012; 24(1):9–15.
[6] Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48(2):124–131.
[7] Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc 2014; 89(4):484–492.
[8] Li J, Wang Q, Zheng W, Ma J, Zhang W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore) 2014; 93(2):100–105.
[9] Sydow M, Schauer A, Crozier TA, Burchardi H. Multiple organ failure in generalized disseminated tuberculosis. Respir Med. 1992; 86(6):517–519.
[10] Mohan A, Sharma SK, Pande JN. Acute respiratory distress syndrome (ARDS) in miliary tuberculosis: a twelve year experience. Indian J Chest Dis Allied Sci. 1996; 38(3):157–162.