Case Report
2017
December
Volume : 5
Issue : 4
Rare case of recurrent Kikuchi-Fujimoto disease
Sarath Chandra Mouli Veeravalli, Suvarna Shilpa S, Satish Rao I, Prathibha B
Pdf Page Numbers :- 135-138
Sarath Chandra Mouli Veeravalli1,*, Suvarna Shilpa S1, Satish Rao I2 and Prathibha B1
1Department of Rheumatology and clinical Immunology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India
2Department of Pathology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India
*Corresponding author: Sarath Chandra Mouli Veeravalli, Clinical Director, Department of Rheumatology and Clinical Immunology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India. Email: sarath10@hotmail.com
Received 15 July 2017; Revised 21 August 2017; Accepted 05 September 2017; Published 15 September 2017
Citation: Veeravalli SCM, SS, Satishrao I, Prathibha B. Rare case of recurrent Kikuchi-Fujimoto disease. J Med Sci Res. 2017; 5(4):135-138. DOI: http://dx.doi.org/10.17727/JMSR.2017/5-25
Copyright: © 2017 Veeravalli SCM, et al. Published by KIMS Foundation and Research Center. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Kikuchi-Fujimoto disease (KFD), or histiocytic necrotizing lymphadenitis, is a rare benign, self-limiting cervical lymphadenitis of unknown etiology. It closely mimics infective and immunological disorders. Here a rare case of recurrent KFD in a 13-year-old girl is presented. This girl presented with low grade fever at night times with sweating of 20 days duration and with painful right cervical lymphadenopathy. She had a bad history of recurrent episodes of lymphadenopathy seven years ago again after two years ago she was treated with empirical ATT. Her routine blood and urine tests were normal. Tests for infection and autoimmunity were negative. Histopathological examination is suggestive of Kikuchi lymphadenitis. Thus, she was diagnosed as a case of recurrent Kikuchi and was treated with oral steroids and hydroxychloroquine (HCQ). To the best of our knowledge, this is the first case of recurrent Kikuchi observed in an Indian patient and treated with Hydroxychloroquine and low dose steroids.
Keywords: Kikuchi-Fujimoto disease; histiocytic necrotizing lymphadenitis; plasmacytoid dendritic cells; hydroxychloroquine
Full Text
Introduction
Kikuchi-Fujimoto disease (KFD) or histiocytic necrotizing lymphadenitis was first described in Japan in 1972 [1, 2]. KFD is a benign disease, characterized by regional cervical lymphadenopathy with tenderness, usually accompanied with mild fever and night sweats. Numerous inciting agents have been proposed, including Epstein-Barr virus (EBV), human herpesvirus 6, human herpesvirus, human immunodeficiency virus (HIV), parvovirus B19, paramyxoviruses, parainfluenza virus, Yersinia enterocolitica, and toxoplasma. Kikuchi syndrome shares sex and age predisposition as well as histologic features with systemic lupus erythematosus (SLE). One ultrastructural study proposed that Kikuchi syndrome reflects a self-limited, SLE-like autoimmune condition caused by virus-infected transformed lymphocytes [3]. There is an exaggerated T-cell-mediated immune response. KFD is generally diagnosed on the basis of an excisional biopsy of affected lymph nodes. Kikuchi’s disease is typically reported to have a low recurrence rate between 3% and 4% [4].
Its incidence has been reported worldwide with a higher prevalence among Japanese and Asiatic individuals. KFD is more common in females compared to males with a male to female ratio of 1:4. People under 30 years of age are more affected by this disease than any other age group [5]. Here a case of recurrent KFD and treated with low dose steroids and HCQ is reported.
Case report
A 13-year-old girl presented, with low grade fever at night times with sweating, for 20 days and with painful right cervical lymphadenopathy. The patient had no symptoms of loss of appetite, weight loss, cough, skin rashes, oral ulcers, alopecia, joint pains or swellings. Her past history revealed that she had right cervical lymphadenopathy seven years ago, for which she was on empirical ATT treatment for a period of 6 months. After that she was asymptomatic for 5 years. Again two years later she had two episodes of prolonged pyrexia associated with axillary lymphadenopathy and she was treated elsewhere with empirical antimalarials for which she responded. Fine needle aspiration cytology (FNAC) of the lymphnode was inconclusive.
Her laboratory investigations were as follows: ESR-13mm/hr, Hb-9.9gm%, WBC count: 6,600 cells/cumm, platelet count: 247000/cumm, MCV: 68.5fl, differential count was neutrophils: 55%, lymphocytes: 38%, eosinophils: 0%, monocytes: 6%, basophils: 1%, angiotensin converting enzyme was normal. Her routine urine test was normal. HBsAg, HCV, HIV I & II, widal, malarial parasite, paul bunnell and mantoux tests were negative. The ANA IF showed weakly positive (1+) in 1:100 titres with pattern Hep 2 cells nucleus dotted and autoantibodies against few dotted nucleus. USG of abdomen and pelvis was normal. USG of neck revealed the presence of multiple conglomerate lymph nodes, the largest node being 18×8 mm with preserved fatty hilum at level-II. Further, the histopathology of the cervical lymph node revealed the presence of large discrete areas of necrosis with abundant nuclear debris surrounded by plenty of histiocytes, transformed lymphocytes and plasmacytoid cells. However no granulocytes, follicular hyperplasia and granulomas were found in the areas of necrosis (Figure 1).
In general, KFD can be differentiated from SLE by using immuno-serologic tests such as ANA, ds-DNA, anti-phospholipid antibodies, C3 and C4 complement tests. Her clinical symptoms did not meet the diagnostic criteria of SLE and clinically no evidence of malignancy or infection was observed. Histopathological examination of lymph node biopsy showed characteristic features similar to lymphadenitis (Figure 1). Further, as the patient presented with recurrent episodes of lymphadenopathy. She was diagnosed as a case of recurrent KFD. She was treated with low dose of steroids and HCQ.
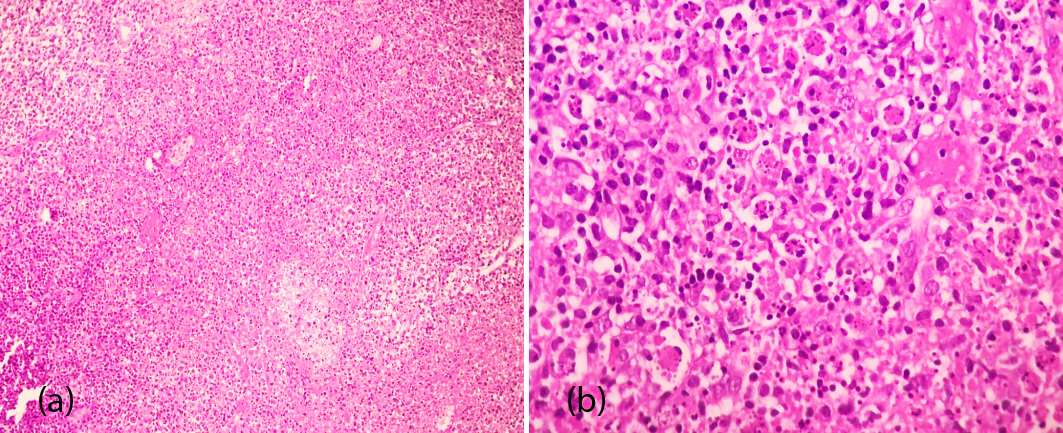
Figure 1: The excisional biopsies of cervical lymph nodes, show (a) Aggregates of histiocytes (H&E stains ×100) and (b) Similar findings consisting of many histiocytes in necrotic background (H&E stains ×400).
Discussion
KFD is a benign disease, characterized by histiocytic necrotizing lymphadenitis in the cervical lymph nodes that affects subjects aged between 20 and 30 years. The common symptoms include fever, fatigue, and cervical lymph node enlargement and the mean duration of symptoms varies between 1 and 4 months [6]. It sometimes presents unusually as axillary and inguinal lymph node enlargement, skin rash, arthralgia, splenomegaly, and aseptic meningitis [6]. KFD can even be complicated with fatal outcomes like disseminated intravascular coagulopathy (DIC), and pulmonary hemorrhage [7].
There are no specific diagnostic methods available for KFD, and its diagnosis is generally by the excisional biopsy of affected lymph nodes. The characteristic histopathologic features of the LN in KFD are the presence of paracortical clusters of plasmacytoid monocytes with interspersed karyorrhexis and crescentic histiocytes. Karryorhectic foci are formed by different cell types, predominantly histiocytes and plasmacytoid monocytes, but also consists of immunoblasts and small and large lymphocytes [8]. Kuo T classified the histopathologic features of KFD into 3 evolving histologic stages: proliferative, necrotizing, and xanthomatous. The proliferative stage consists basically of various histiocytes, plasmacytoid monocytes, and a variable number of lymphoid cells with karyorrhectic nuclear fragments and eosinophilic apoptosis debris. If cellular aggregates in a given lymph node showed any degree of coagulative necrosis, the case was classified as necrotizing. If foamy histiocytes predominated in the KFD lesions, the case was classified as xanthomatous regardless of the presence or absence of necrosis [5].
The lymphocytes within and surrounding the typical areas of necrosis in affected lymph nodes of KFD is characterized by nuclear fragmentation, a typical feature of early apoptosis. Mainly CD8+ cytotoxic T cells undergo apoptosis, and proliferation in KFD [9]. The high Fas/FasL (ligand) among CD8+ T cells rather than CD4+ T cells would probably lead to apoptosis in the CD8+ T cells. Further, CD123+ plasmacytoid predendritic cells—the so called plasmacytoid monocytes or plasmacytoid T cells have been reported to be a striking histopathologic finding of KFD. These cells might produce large amounts of type I interferon, which promotes a T-helper 1 T-cell response and induces cytotoxic immune reaction leading to apoptosis. Accumulation of apoptotic plasmacytoid monocytes in KFD parallels defective clearance of apoptotic cells in SLE and thus supports a pathophysiologic association between KFD and SLE [10].
Most of the affected patients recover without serious complications, although some of them suffer from recurrent episodes of Kikuchi’s disease. A relatively low recurrence rate has been described in 3-4% of cases [4]. The site of recurrence is usually the same as the first site. Comparison of European cases and cases from Asia, Asian patients are reported to have higher ANA positivity than Europeans. ANA positivity is a risk factor for recurrent KFD and KFD associated with SLE [11].
Patients with classic symptoms are treated with NSAIDs or steroid, and those with severe symptoms are given high dose steroids and IVIG. Immunosuppressant is recommended for complicated cases to prevent fatal outcome [12]. Rezai et al. reported the first use of HCQ to treat recurrent KFD [13].
HCQ is a basic 4-aminoquinolone compound generally used as an antimalarial agent. There are several mechanisms by which HCQ mediates its action. HCQ is involved in immunomodulation, induction of apoptosis, lymphoid accumulation, and posttranslational viral glycosylation [14]. It is shown to inhibit several enzymes and interfere with monocyte-macrophage and suppresses the production of proinflammatory cytokines, such as interleukin (IL)-1, IL-6 at posttranscriptional level. HCQ induces apoptosis of effector T cells by inhibiting autophagy [15], and inhibit T cell antigen receptor signaling pathway through the calcium signal [16]. Further, HCQ makes alkalization of acidic intracellular vesicles, required for endosomal toll-like receptor activation and thus results in decreased inflammatory cytokine production and antigen processing necessary for antigen presentation of autoantigens. Few reports have showed the effectiveness of hydroxychloroquine (HCQ) to treat recurrent KFD [17-19].
Similarly, in the present case, low dose steroids and HCQ were used to treat this patient. Subsequently the patient recovered and she has been on regular follow-up. Our findings in the present case support the efficacy of the concomitant use of HCQ along with low dose corticosteroid for recurrent KFD patients.
Conflict of interest
Authors declare no conflict of interest.
References
[1] Kikuchi M. Lymphadenitis showing focal reticulum cells hyperplasia with nuclear debris and phagocytosis: A clinicopathological study. Acta Hematol Jpn. 1972; 35:379–380.
[2] Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis. Naika 1972; 30:920–927.
[3] Imamura M, Ueno H, Matsuura A, Kamiya H, Suzuki T, et al. An ultrastructural study of sub acute necrotizing lymphadenitis. Am J Pathol. 1982; 107(3):292–299.
[4] Song JY, Lee J, Park DW, Sohn JW, Suh SI, et al., Clinical outcome and predictive factors of recurrence among patients with Kikuchi's disease. Int J Infect Dis. 2009; 13(3):322–326.
[5] Kuo T. Kikuchi's disease (histiocytic necrotizing lymphadenitis): a clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology and DNA ploidy. Am J Surg Pathol. 1995; 19(7):798–809.
[6] Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004; 122(1):141–152.
[7] Uslu E, Gurbuz S, Erden A, Aykas F, Karagoz H, et al., Disseminated intravascular coagulopathy caused by Kikuchi–Fujimoto disease resulting in death: first case report in Turkey. Int Med Case Rep J. 2014; 7:19–22.
[8] Tsang WY, Chan JK, Ng CS. Kikuchi's lymphadenitis. A morphologic analysis of 75 cases with special reference to unusual features. Am J Surg Pathol. 1994; 18(3):219–231.
[9] Ohshima K, Haraoka S, Takahata Y, Takada H, Tsutiya K, et al., Interferon-gamma, interleukin-18, monokine induced by interferon-gamma and interferon-gamma-inducible protein-10 in histiocytic necrotizing lymphadenitis. Leuk Lymphoma. 2002 43(5):1115–1120.
[10] Facchetti F, Vermi W, Mason D, Colonna M. The plasmacytoid monocyte/interferon producing cells. Virchows Arch. 2003; 443(6):703–717.
[11] Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007; 26(1):50–54.
[12] Lin DY, Villegas MS, Tan PL, Wang S, Shek LP. Severe Kikuchi's disease responsive to immune modulation. Singapore Med J. 2010; 51(1):e18–21.
[13] Rezai K, Kuchipudi S, Chundi V, Ariga R, Loew J, et al. Kikuchi-Fujimoto disease: hydroxychloroquine as a treatment. Clin Infect Dis. 2004; 39(12):e124–e126.
[14] Olsen NJ, Schleich MA, Karp DR. Multifaceted effects of hydroxychloroquine in human disease. Semin Arthritis Rheum. 2013; 43(2):264–272.
[15] Van Loosdregt J, Spreafico R, Rossetti M, Prakken BJ, Lotz M, Albani S. Hydroxychloroquine preferentially induces apoptosis of CD45RO+ effector T cells by inhibiting autophagy: a possible mechanism for therapeutic modulation of T cells. J Allergy Clin Immunol. 2013; 131(5):e1443–1446.
[16] Goldman FD, Gilman AL, Hollenback C, Kato RM, Premack BA, et al. Hydroxychloroquine inhibits calcium signals in T cells: a new mechanism to explain its immunomodulatory properties. Blood. 2000; 95(11):3460–3466.
[17] Chen PH, Huang YF, Tang CW, Wann SR, Chang HT. KikuchiFujimoto disease: an amazing response to hydroxychloroquine. Eur J Pediatr 2010; 169(12):1557–1559.
[18] Hyun M, So IT, Kim HA, Jung H, Ryu SY. Recurrent Kikuchi’s disease treated by hydroxychloroquine. Infect Chemother. 2016; 48(2):127–131.
[19] Fumika H, Hiroto T, Hirofumi T, Ayako O, Hidenori T, et al., Recurrent Kikuchi-Fujimoto Disease Successfully Treated by the Concomitant Use of Hydroxychloroquine and Corticosteroids. Intern Med. 2017; 56(24):3373–3377.