Full Text
Case 1 (Oncogenic osteomalacia)
Hypophosphatemic phosphaturic oncogenic osteomalacia is an acquired paraneoplastic disease, first recognized in 1947 [1]. It is characterized by hypophosphatemia, phosphaturia, normocalcemia, and osteomalacia in the absence of a nutritional or drug history suggestive of vitamin D deficiency or generalized renal tubular defects. Hypophosphatemia occurs as a result of excessive renal phosphate excretion secondary to various types of tumors usually benign mesenchymal tumors, others include fibrous dysplasia, osteosarcoma, hemangiopericytoma, chondroblastoma, chondromyxoid fibroma, malignant fibrous histiocytoma, giant cell tumour, haemangioma, paraganglioma, prostate cancer and oat cell carcinoma of the lung. Patients typically present with a history of chronic bone pain, fractures, and proximal motor weakness. Children may exhibit poor growth and lower extremity deformity. A recent report has suggested that fibroblast growth factor 23 (FGF-23) is the most reliable marker for the detection of these tumors [2].
A case of tumor-induced osteomalacia associated with an occipital bone tumor is reported here. The tumor was successfully resected in toto. Phosphate level recovered to normal range after the surgery and patient also symptomatically improved.
Case presentation
56-years-old male who was a known diabetic for 12 years presented with history of progressive weakness of all four limbs with pains and tremors for 3 years, the pains and weakness progressively worsened in 9 months, eventually patient became wheel chair bound, without sensory involvement or bladder bowel disturbances. Patient noticed swelling in his occipital region which was painless and slowly increasing in size for two and half years. Patient had history of fracture of left femural neck 2 years ago, and had been operated for the same. On examination patient was moderately built with hypotonia of all 4 limbs and flabby muscles, fine tremors on outstretched hands were present. Power in all four limbs 4+/5 with proximal muscle weakness, with preserved sensory and autonomic functions. Local examination revealed 8*5 cm swelling in the occipital region which was diffuse, firm to hard in consistency with a scar. It was not pulsatile, not tender and cough impulse was absent. Patient was evaluated by a neuro physician initially and MRI spine was done which revealed C4-C5, L4-L5, L5-S1 disc prolapse without any significant canal stenosis or cord compression. On biochemical analysis patient was found to have hypophosphatemia due to renal loss of phosphate with the Tubular maximum reabsorption rate of phosphate (TMP)/ Glomerular filtration rate (GFR)=1. Serum calcium was low with elevated parathormone. Compound muscle action potentials (CMAPS), sensory nerve action potentials (SNAPS), electromyography (EMG) were suggestive of primary muscle disease. Patient was managed with intravenous calcium, vitamin D3, oral phosphate and oral calcium. Patient was subjected for incisional biopsy of occipital swelling which revealed phosphaturic mesenchymal tumor of occipital bone. Patient was further evaluated with whole body positron emission tomography (PET), and magnetic resonance imaging (MRI) brain. In PET scan a hot spot was noted on the occipital region which was positive for somatostatin receptor. MRI revealed heterogeneous bony mass lesion arising from the occipital bone left side. It was isointense on T1, heterogeneous mixed intensity signals on T2, mild to moderate heterogeneous enhancement with contrast, with compression of ipsilateral transverse sinus, with normal brain parenchyma.
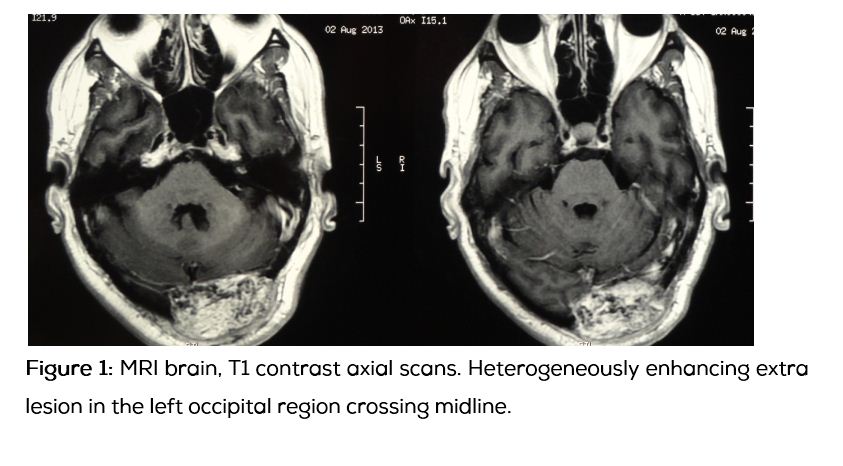
Table 1: Biochemical parameters before and after surgery.
|
Serum level
|
Pre-operative
|
Post-operative(at 3 months )
|
Biological reference
|
|
Fasting calcium
|
6.9 mg/dL
|
8.9 mg/dL
|
8.3 - 10.4 mg/dL
|
|
Fasting phosphate
|
1.9 mg/dL
|
4.0 mg/dL
|
3.5 - 5.0 mg/dL
|
|
Alkaline phosphatase
|
410 U/L
|
245 U/L
|
32 _ 91 U/L
|
|
Serum albumin
|
3.8 mg/dL
|
3.8 mg/dL
|
|
|
Creatinine
|
1.5mg/dL
|
1.2mg/dL
|
0.5 - 1.4 mg/dL
|
|
Fasting blood glucose
|
132 mg/dL
|
156 mg/dL
|
|
|
Post-prandial glucose
|
190 mg/dL
|
201 mg/dL
|
|
|
Sodium
|
139 mg/dL
|
137 mg/dL
|
135 - 145 mg/dL
|
|
Potassium
|
4.1 mg/dL
|
4.0 mg/dL
|
3.5 - 5.0 mg/dL
|
|
Bicarbonate
|
25 mg/dL
|
26 mg/dL
|
22 -29 mg/dL
|
|
Chloride
|
104 mg/dL
|
102 mg/dL
|
95 - 105 mg/dL
|
|
Spot urine phosphate
|
17.48mg/dL
|
11.48mg/dL
|
|
|
Spot urine creatinine
|
216.39mg/dL
|
216.39mg/dL
|
|
|
TMP/GFR
|
1
|
|
|
|
25 (OH) vitamin D
|
38.1ng/ml
|
42.1ng/ml
|
30 _100 ng/ml
|
|
Plasma intact PTH
|
114.5 pg/ml
|
92.5 pg/ml
|
12-88 pg/ml
|
|
GFR
|
51.45
|
58.00
|
>=60
|
Patient underwent excision of the occipital lesion, intra operatively tumor was involving the entire thickness of the bone. The normal bone and the tumor margins were well delineated by drilling the bone all around the tumor and the normal dura visualized. Tumor was removed piecemeal. Intra operatively tumor was gritty, highly vascular, eroding bone, easily separable from dura without any dural invasion or sinus involvement.
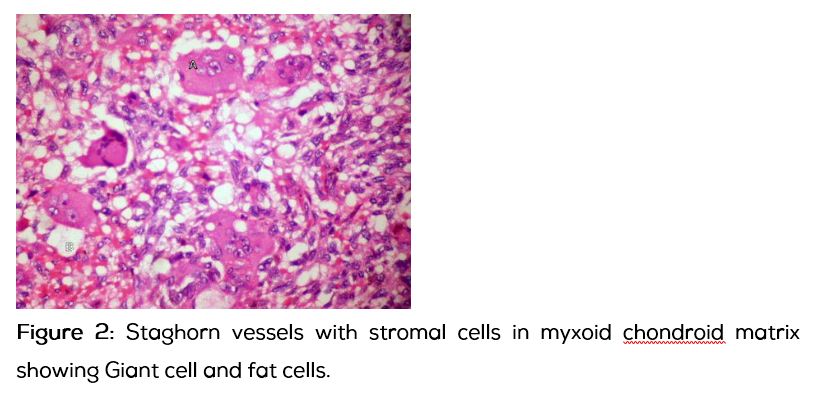
Histopathological examination (HPE): Tumor composed of round to spindle cells in a myxoid chondroid matrix interspersed by mature fat and osteoid matrix. The vascularity was prominent with focal staghorn vessels. Many osteoclastic giant cells were noted haphazardly arranged. Mitosis was <1/10HPF. Large hemosiderin laden macrophages were noted. Features suggestive of phosphaturic mesenchymal tumor of mixed connective tissue type.
In the post-operative period, after three months follow up patients serum calcium and phosphate returned to normal level but serum alkaline phosphatase and parathormone levels were still high and symptomatically patient’s condition improved to a level that patient could walk for 20 footsteps independently.
Discussion
Oncogenic osteomalacia is an unusual condition, but it probably still is the most common cause for acquired hypophosphatemic osteomalacia in adult males. The manifestations are similar to those of hereditary phosphate wasting disorders and the primary event is induction of severe phosphaturia by a humoral factor secreted by the tumour. Tumors secrete fibroblast growth factor 23 (FGF-23), which inactivates sodium phosphate cotransporters in the proximal renal tubule which results in reduced phosphate reabsorption in the nephron, causing severe hypophosphatemia [3]. FGF-23 also reduces the expression of 25-hydroxy-1α-hydroxylase, which is needed for the conversion of inactive vitamin D to its active form. Thus, patients have low serum phosphate levels and also low or normal serum 1,25-dihydroxy vitamin D levels [4]. Since active vitamin D promotes intestinal phosphate absorption, patients with oncogenic osteomalacia have difficulty absorbing phosphate from their diet also.
Mutations in fibroblast growth factor 23 (FGF-23) cause autosomal dominant hypophosphatemic rickets. Clinical and laboratory findings in this disorder are similar to those in oncogenic osteomalacia, in which tumors abundantly express FGF-23 messenger RNA, and to those with X-linked hypophosphatemia, which is caused by inactivating mutations in a phosphateregulating endopeptidase. Recombinant FGF-23 induces phosphaturia and hypophosphatemia in vivo, suggesting that it has a role in phosphate regulation. FGF-23 is readily detectable in the plasma or serum of healthy persons and can be markedly elevated in those with oncogenic osteomalacia or X-linked hypophosphatemia, suggesting that this growth factor has a role in phosphate homeostasis. FGF-23 measurements might improve the management of oncogenic osteomalacia [5].
Patients experience poor bone mineralization and a high frequency of fractures of the long bones, ribs, and vertebrae. Other symptoms include severe bone pain and muscle weakness. Our patient also had fracture left femur with severe bone pains. These tumors are benign in nature. Even in histologically malignant tumors, local recurrence or distant metastasis is extremely rare. The most common types of these tumors are hemangiopericytomas. Other types include fibromas, chondrosarcomas, neuroblastomas, and prostate carcinomas [6, 7]. Somatostatin analog imaging may aid in localization, because of the presence of such receptors in some of these tumors [8].
The treatment of choice is surgical removal of the tumour. There is a dramatic improvement in the clinical course of oncogenic osteomalacia when the offending tumour is correctly identified and completely removed. With the half-life of FGF-23 being approximately an hour, post-surgery FGF-23 levels fall off drastically [9, 10]. Serum phosphate level and TMP/ GFR of phosphate return to normal within hours to days after the removal of the tumor. 1,25-dihydroxy vitamin D levels return to normal within days, while long-term skeletal changes reverse within months [10]. However clinical resolution of symptoms and serum biochemical markers of bone turnover, such as the osteocalcin level and alkaline phosphatase activity, tend to take longer to normalize. Therapy may be aided by short term replacement of phosphate orally and addition of Vitamin D and calcium. Our patient also showed change in biochemical parameters after excision of the tumor, serum calcium and phosphate levels came to normal but serum alkaline phosphatase and parathormone levels were still high after three months. Postoperative FGF-23 levels also may be used to determine the adequacy of tumor resection [9]. Unfortunately FGF-23 tumor markers are not available in this institute and it was not done in this patient. Tumor size and location may result in incomplete resection or adjacent soft-tissue damage, since the lesion may not have a distinct boundary from surrounding tissue. Therefore, other techniques, such as radiofrequency ablation, may be used when surgery is not possible [11]. This patient had good recovery after the surgical excision of the tumor.
Case 2: Aneurysmal bone cyst of the calvaria
Introduction
Aneurysmal bone cyst is a rare benign vascular lesion, considered secondary to certain pathological conditions. Aneurysmal bone cysts are common in long bones and rare to occur in calvarium. A 13-year-old boy presented with aneurysmal bone cyst of the right parietal bone who underwent total excision and cranioplasty is reported.
Case presentation
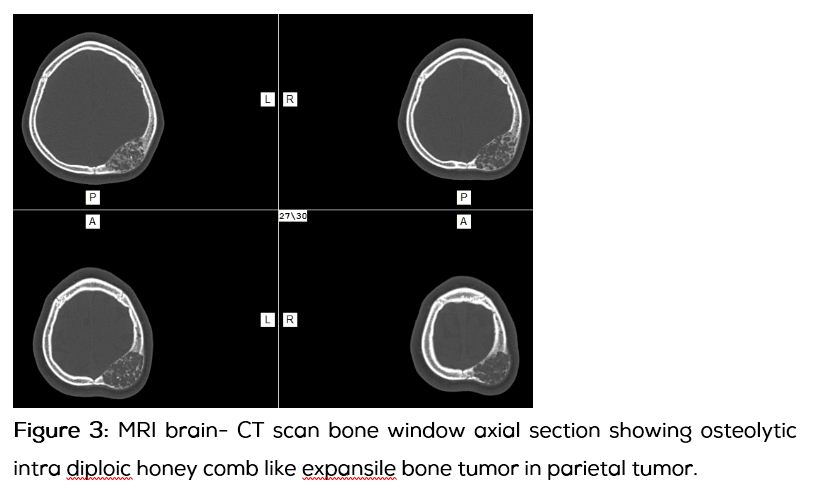
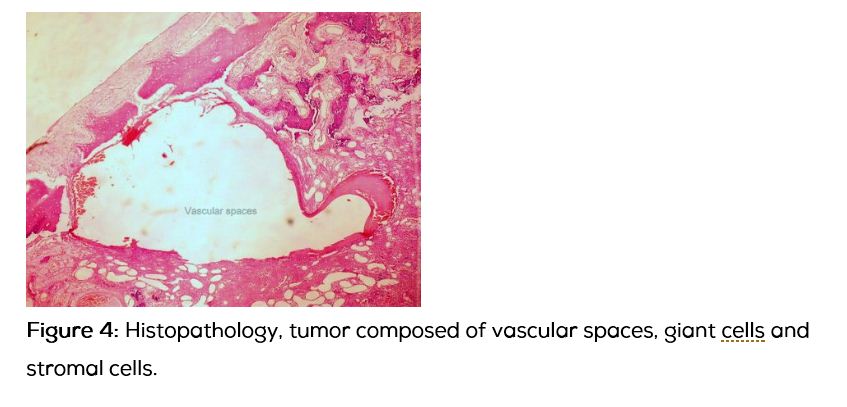
13-year-old boy presented with progressively increasing size of the swelling in the left parietal region over 3 years without any pain. Apart from the swelling the child was asymptomatic. There was no significant past history. On examination 8x8 cm swelling with diffuse borders, bony hard consistency, non-pulsatile, with absent cough impulse, without any venus hum or bruit noted over the left parietal region. Computed tomography of the skull and brain showed fusiform shaped expansile osteolytic intra diploic lesion which had typical soap bubble or honey comb like appearance with intra diploic trabeculae which was suggestive of aneurysmal bone cyst. Brain parenchyma was normal except for the minimal cortical buckling of the ipsilateral parietal cortex. Patient underwent surgical excision of the tumor en bloc with a margin of 1cm all around along with cranioplasty. Intra operatively, the involved bone was ballooned out with bluish hue on the surface with lot of branching vessels, outer and inner tables were thin and easily breakable, without any dural involvement. Histopathologically the tumor showed evidences of large blood filled spaces surrounded by stromal and scattered giant cells, which was suggestive of primary aneurysmal bone cyst.
Discussion
Calvarial aneurysmal bone cyst is a rare entity. Few cases have been reported in literature. These lesions occur most commonly in patients under 20 years old of both genders [12]. Radiographs reveal an eccentric, lytic lesion typically with an expanded, remodeled “blown-out” or “ballooned” bony contour of the affected bone, with a delicate trabeculated appearance frequently [13]. It may be a primary aneurysmal bone cyst or it can occur as a result of a preexisting pathological condition, such as Giant Cell Tumours (GCTs), fibrous dysplasia, non-ossifying fibromas, haemangiomas, osteoblastomas, simple bone cysts, chondroblastomas, chondromyxoid fibromas and even osteosarcomas. An interesting theory about the aetiology of primary ABCs is that the lesions occur due to haemorrhage in the bone as a result of increased venous pressure. The haemorrhage is thought to lead to osteolysis. The osteolysis, in turn, causes further haemorrhage, leading to exponential growth of the tumour [14]. This theory would perhaps explain why aneurysmal bone cysts (ABC) are uncommon in the calvarium and bones of the facial skeleton, where the venous pressure is low. On the other hand, ABCs are common in long bones, where the venous pressure is high and the marrow content is greater. As ABCs are known to recur, the optimal treatment is total excision. Other established treatment modalities, especially in complex areas such as the skull base, include arterial embolization, injection sclerotherapy, curettage (with or without bone grafting), cryotherapy, aspiration and drainage, radionuclide ablation, radiotherapy or a combination of these modalities. Radiotherapy alone, however has recently fallen out of favour owing to the risk of post-irradiation sarcoma [15]. In secondary ABCs, the treatment plan is formulated according to the primary lesion that is identified. For example, an ABC in an osteosarcoma is treated more aggressively than an ABC that has developed in pre-existing fibrous dysplasia.
Conclusion
Hypophosphatemic mesenchymal tumor is a rare entity causing adult onset oncogenic osteomalacia, occurrence of such tumor in calvarium is still more rare. It is a benign tumor and complete excision of tumor will revert the serum biochemical level of phosphate and calcium in the patient. Aneurysmal bone cysts are also rare vascular tumors of skull bones and may occur as a primary ABC or secondary to preexisting pathology. Treatment might vary from simple excision to combinations of therapy depending upon its type.
Acknowledgements
Acknowledgements are due to Department of Radiology and Imaging, KIMS, Secunderabad, Telangana, India.
Conflict of interest
The authors declare no conflict of interest.
References
1. McCance RA. Osteomalacia with Looser’s zones (Milkman’s syndrome) due to raised resistance to vitamin D acquired about the age of fifteen years. Q J Med. 1947; 16(1):33–47.
2. Jan de Beur SM. Tumor-induced osteomalacia. JAMA. 2005; 294(10):1260–1267.
3. Jan de Beur SM. Tumor-induced osteomalacia. JAMA 2005; 294 (10):1260–1267.
4. Carpenter TO. Oncogenic osteomalacia: a complex dance of factors. N Engl J Med. 2003; 348 (17):1705–1708.
5. Bielesz B, Klaushofer K, Oberbauer R. Renal phosphate loss in hereditary and acquired disorders of bone mineralization. Bone. 2004; 35(6):1229–1239.
6. Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004, 28(1):1–30.
7. Kumar R. Tumor-induced osteomalacia and the regulation of phosphate homeostasis. Bone. 2000; 27(3):333–338.
8. Jan de Beur SM, Streeten EA, Civelek AC, McCarthy EF, Uribe L, et al. Localisation of mesenchymal tumours by somatostatin receptor imaging. Lancet. 2002; 359(9308):761–763.
9. Carpenter TO. Oncogenic osteomalacia: a complex dance of factors. N Engl J Med. 2003; 348(17):1705–1708.
10. Khosravi A, Cutler CM, Kelly MH, Chang R, Royal RE, et al. Determination of the elimination half-life of fibroblast growth factor-23. J Clin Endocrinol Metab. 2007; 92(6):2374–2377.
11. Hesse E, Rosenthal H, Bastian L. Radiofrequency ablation of a tumor causing oncogenic osteomalacia . N Engl J Med. 2007; 357(4):422–424.
12. Składzień J, Oleś K, Zagólski O, Moskała M, Sztuka M, et al. A giant cranial aneurysmal bone cyst associated with fibrous dysplasia. B-ENT. 2008; 4(1):29–33.
13. Kransdorf MJ, Sweet DE. Aneurysmal bone cyst: concept, controversy, clinical presentation and imaging. AJR Am J Roentgenol. 1995; 164(3):573–580.
14. Lin WC, Wu HT, Wei CJ, Chang CY. Aneurysmal bone cyst arising from fibrous dysplasia of the frontal bone (2004:2b). Eur Radiol. 2004; 14(5):930–932.
15. Kamikonya N, Hishikawa Y, Kurisu K, Taniguchi M, Miura T. Aneurysmal bone cyst treated by high-energy, low-dose radiation therapy: a case report. Radiat Med. 1991; 9(2):54–56.