Case Report
2015
June
Volume : 3
Issue : 2
Apert syndrome
Begum Azra, Sandeep, Ananta Ram Gudipati, Rajinikanth Rao V
Pdf Page Numbers :- 70-74
Dr. Begum Azra1,*, Dr. Sandeep1, Dr. Ananta Ram Gudipati1 and Dr. Rajinikanth Rao V1
1Radiology and Imageology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India
*Corresponding author: Dr. Azra Begum, Radiology and Imageology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India.
Received 10 January 2015; Revised 9 March 2015; Accepted 18 March 2015; Published 25 March 2015
Citation: Begum A, Sandeep, Ananta Ram G, Rajinikanth Rao V. Apert syndrome. J Med Sci Res. 2015; 3(2):70-74. DOI: http://dx.doi.org/10.17727/JMSR.2015/3-013
Copyright: © 2015 Begum A, et al. Published by KIMS Foundation and Research Center. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Craniosynostosis is due to premature closure of the cranial sutures. The radiological findings of a 3-year-old girl with a small head, triangular frontal shape, hypertelorism, delayed milestones and syndactyly are presented. A provisional diagnosis of Apert syndrome was made and radiological investigations were performed. Radiographs of both hands and feet showed soft tissue fusion of all digits and synostosis involving the phalanges of both hands and second and third metacarpals of both feet. Radiograph of the skull showed craniosynostosis with turribrachycephaly. Three-dimensional computed tomography (CT) revealed a sagittal, coronal and metopic suture synostosis with exaggerated convolutional markings. MRI of the brain demonstrated agenesis of corpus callosum. Diffusion tensor MRI and fiber tractography (FT) were performed which showed white matter fiber orientation and neuroanatomic configuration of aberrant hemispheric fiber connections.
Keywords: Apert syndrome; syndactyly; craniosynostosis
Full Text
Apert syndrome is a rare congenital disorder which is characterised by craniosynostosis, syndactyly of the hands and feet with a tendency for fusion of osseous structures. Craniosynostosis syndrome is defined as premature closure of the cranial sutures producing deformity of the skull which includes Crouzon’s syndrome, Pfieffer's syndrome, Apert syndrome, Jackson- Weiss syndrome [1]. A case of Apert syndrome highlighting the role of 3D computed tomography (CT), magnetic resonance imaging (MRI) and radiographs in the diagnosis and management is reported. In this article, a novel technique is described, diffusion MR imaging and fiber tractography (FT), which shows the white matter fiber orientation in vivo, and neuroanatomic configuration of aberrant hemispheric fiber connection in agenesis of corpus callosum.
Case presentation
A 3-year-old girl with a small cranium, triangular forehead and symmetrical syndactyly of hands and feet presented to KIMS Hospital (Figure 1). She had a normal uneventful full term delivery, born out of a non-consanguineous marriage. She had an elder sibling who was completely normal. On physical examination, both anterior and posterior fontanelle were closed. The contour of the skull was abnormal (turribrachycephaly). She showed mid face hypoplasia, hypertelorism, proptosis and a high arched palate. Based on these findings, a provisional diagnosis of apert syndrome was made. Further radiological investigations were performed.
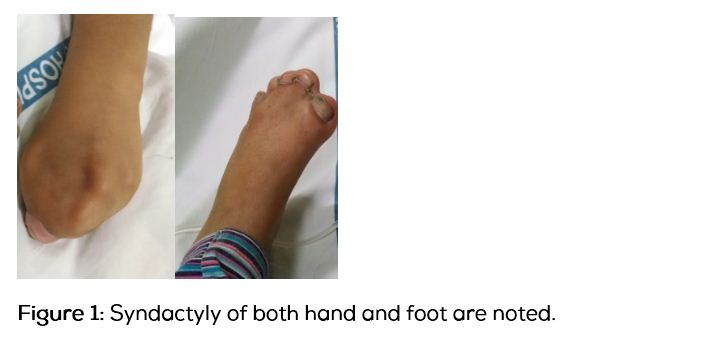
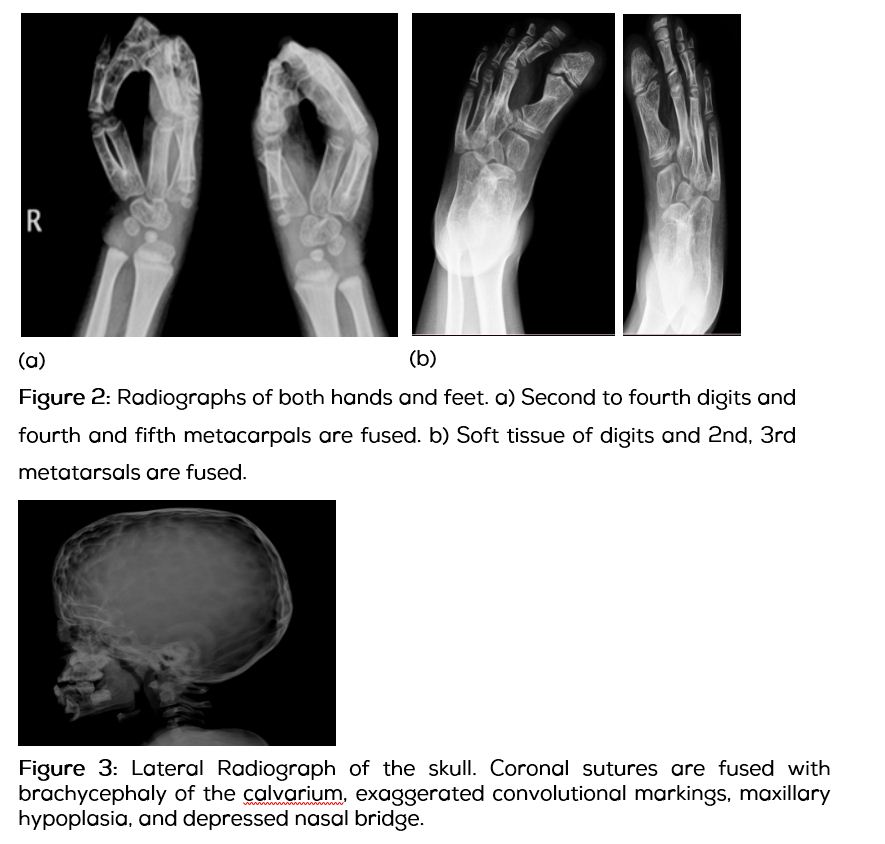
Radiographs of both hands and feet demonstrated soft tissue syndactyly of all digits and synostosis involving the phalanges of first, second, third and fourth digits. Synostosis of fourth and fifth metacarpals of both hands was noted. Both feet showed soft tissue fusion of all digits and synostosis of the second and third metatarsals of both feet with deformed phalanges (Figure 2). Skull radiographs revealed fused coronal, sagittal and lamboid sutures, turribrachycephaly and triangular forehead with prominent convolutional markings and widened sella turcica suggestive of increased intracranial pressure (Figure 3).
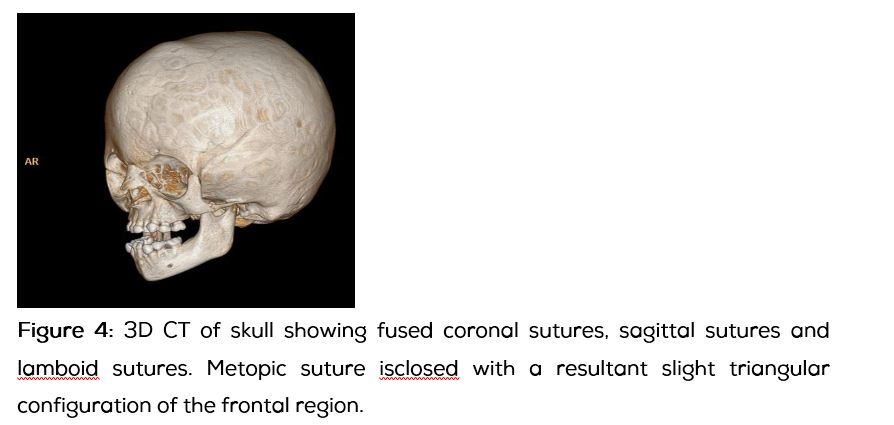
CT of the brain showed fused sutures and turribrachicephaly. Expansion of the middle cranial fossa, elevation and forward bowing of the sphenoid ridges, lateral bowing of the temporal squamosa and marked scalloping of the inner table were observed. Volume rendered images of the cranium showed multiple thinned areas in the cranial vault, convexities of which were facing outwards and focal defects in the posterior parietal and occipital regions due to exaggerated convolutional gyri. Increased predental interval and mild retroflexion of dens tip was noted in the cranio-vertebral junction. Fused coronal sutures, sagittal sutures and lamboid sutures were seen. Metopic suture is closed with a resultant slight triangular configuration of the frontal region (Figure 4).
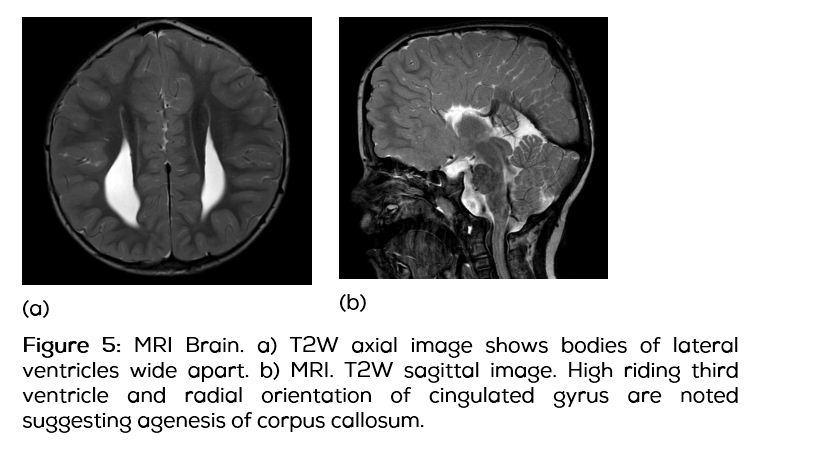
Further imaging with MRI revealed parallel bodies of lateral ventricles and pointed and pinched out frontal horns. Complete agenesis of corpus callosum with radial orientation of cingulate gyri and a high riding third ventricle were noted (Figure 5). Prominent temporal horns of both lateral ventricles and Probst bundles bilaterally and vertical orientation of hippocampi were seen.
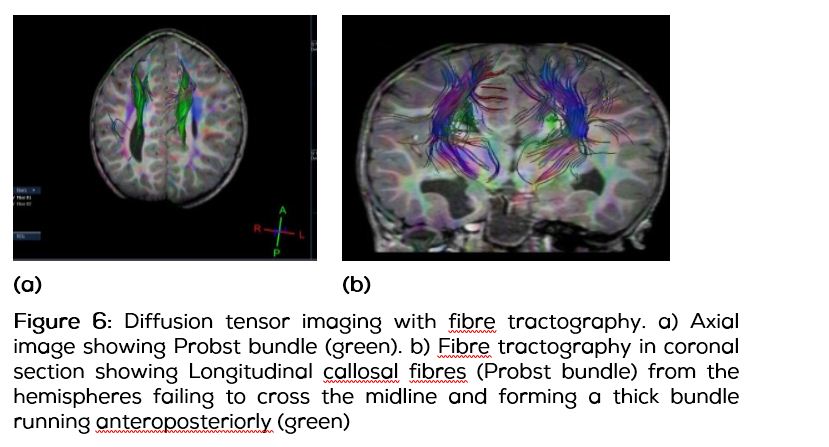
Diffusion tensor imaging was performed by using a 3-T system (Philips Medical System) with synergy-L sensitivity encoding (SENSE) head coil. Diffusion weighting was performed by using single-shot spin-echo echo planar imaging and navigator echo phase correction. Parameters of diffusion tensor imaging were as follows: a data matrix of 112 over a 23-cm field of view, 5mm section thickness with a gap of 1.5 mm, TE = 80 ms, TR = 2153ms, SENSE factor = 2; number of acquisitions = 20, b = 1000 s/mm2 with six different directions, and total imaging time less than 1 minute. Anisotropy was calculated by using the orientation-independent fractional anisotropy (FA) and diffusion tensor color maps were created from FA values and the three vector elements. Vector maps were assigned to red (x element, left-right), green (y, anteroposterior), and blue (z, superior-inferior) with proportional intensity scale according to the FA. Three-dimensional FT was obtained. Fiber bundles were coded as 2D color maps (eg, red [x], green [y], and blue [z]). In the case of complete agenesis of corpus callosum, Longitudinal callosal fibres (LCF) fibers from the hemispheres failed to cross the midline and formed a thick bundle running anteroposteriorly (Probst bundle), which showed as a dark white matter tract on T2-weighted images and as a thick bundle of clear green color on diffusion tensor-based color map (Figure 6).
Discussion
Dr. Eugene Apert in 1906, described a syndrome as characterized by the clinical trial of craniosynostosis, midface hypoplasia, and symmetric syndactyly of the hands and feet [2]. The prevalence of Apert syndrome has been calculated to be approximately 15.5 per million live births [3].
The etiology is unclear but various theories have been proposed [4-6]. The most favoured theory is still based on that originally proposed by Virchow in 1856 which describes intrinsic abnormality of the sutures resulting in fusion and a reduction in growth perpendicular to the plane of the suture1. Inherited in an autosomal dominant fashion, most cases arise as spontaneous mutations that appear to originate almost exclusively in the paternal germ line. Two mutations on chromosome10q, found in adjacent codons leading to altered structure in the fibroblast growth factor receptor (FGFR 2) have been identified as being responsible for the defects seen in Apert syndrome. Almost 80% of the cases of crainiosynostosis arise sporadically rather than being associated with syndromes. The two most common of the syndromic craniosynostoses, collectively known as acrocephalosyndactyly, are Crouzon's and Apert, which together make up 70% of such cases [7].
Apert syndrome has distinctive clinical features. The coronal suture fuses prematurely (at less than 3 months), leading to an acrocephalic (cone-shaped) head with shortened antero-posterior diameter, and a high prominent forehead [8-11]. In our case, in addition to the coronal suture, the sagittal, lambdoid and metopic sutures were also fused. This is a unique feature in our case, as it has not been reported previously. Brachycephaly, hypertelorism, midface hypoplasia occurs. There is prominence of the mandible with resultant malocclusion. These features were present in our case. A high arched palate and cleft palate (30%) is a feature [1]. Our case had a high arched palate, however there was no associated cleft. The digital manifestations include distal (acro-) fusion (syndactyly) of commonly the second to fourth digits of the hands. These findings are frequently symmetrical. The thumb may be incorporated into the mid-hand syndactyly as seen in our patient. Progressive longitudinal fusions also occur (symphalangism). Fusion of bodies of C5 and C6 are seen in our patient, a common feature noted in 70% of the cases [1]. There are associated cardiac and genitourinary abnormalities in 10% of patients. Ventriculomegaly and abnormalities of the corpus callosum and septum pellucidum are not uncommon. Examination did not reveal any cardiac or genitourinary abnormalities. MRI showed ventriculomegaly with agenesis of corpus callosum, absent septum pellucidum, vertical hippocampi and a high riding third ventricle. Previous studies reported affected individuals with anomalies of viscera, elbows and shoulders, skeleton and central nervous system with impaired mental function [10, 12]. There were no anomalies of the viscera, elbows or shoulders.
Apert syndrome is a rare disorder, however it should be recognized early, as neonatal diagnosis can be attempted via genetic workup on the basis of suspicious sonographic findings.
Mothers may be offered genetic counseling during pregnancy. Many surgical treatment options can produce better outcome if performed early. Craniotomy within the first year of life has been found to result in higher adult intelligence levels, with 50% of such patients having an IQ ≥70, as compared with only 7% of those patients in whom such a procedure is performed later in life [15].
Conclusion
Diffusion tensor imaging and fibre tractography clearly demonstrated the corpus callosum agenesis. This is an additional feature in this case report which has not been demonstrated in previous studies. The use of CT with 3D volume reconstruction has helped considerably in the precise diagnosis of craniosynostosis as the sutures were identified more clearly in CT than on radiographs for planning surgical reconstruction.
Acknowledgements
Acknowledgements are due to the Krishna Institute of Medical Sciences (KIMS), Minister Road, Secunderabad – 500003, Telangana, India.
Conflict of Interest
The authors declare no conflict of interest.
References
1. Aviv RI, Rodger E, Hall CM. Craniosynostosis. Clin Radiol. 2002; 57(2):93–102.
2. Apert E. De l'acrocephalosyndactylie. Bull Soc Med Paris. 1906; 23:1310–1330.
3. Cohen MM Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992; 42(5):655–659.
4. Marsh JL, Vannier MW. Cranial base changes following surgical treatment of craniosynostosis. Cleft Palate J. 1986; 23 Suppl 1:9–18.
5. Moss ML. The pathogenesis of premature cranial synostosis in man. Acta Anat (Basel). 1959; 37:351–370.
6. Muenke M, Schell U, Hehr A, Robin NH, Losken HW, et al. A common mutation in the fibroblast growth factor receptor gene in Pfieffer syndrome. Nat Genet. 1994; 8(3):269–274.
7. Munke M, Schell U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 1995; 11:308–313.
8. DeGiovanni CV, Jong C, Woollons A. What syndrome is this? Apert syndrome. Pediatric Dermatol. 2007; 24:186–188.
9. Freiman A, Tessler O, Barankin B. Apert syndrome. Int J Dermatol. 2006; 45(11):1341–1343.
10. Surman TL, Logan RM, Townsend GC, Anderson PJ. Oral features in Apert syndrome: a histological investigation. Orthod Craniofac Res. 2010; 13(10):61–67.
11. Albuquerque MA, Cavalcanti MG. Computed tomography assessment of Apert syndrome. Braz Oral Res. 2004; 18(1):35–39.
12. Carneiro GV, Farias JG, Santos FA, Lamberti PL. Apert syndrome: review and report a case. Braz J Otorhinolaryngol. 2008; 74(4):640.
13. Letra A, de Almeida AL, Kaizer R, Esper LA, Sgarbosa S, et al. Intraoral features of Apert’s syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007; 103(5):e38– e41.
14. Verma S, Draznin M. Apert syndrome. Dermatol Online J. 2005;11:15.
15. Renier D, Arnaud E, Cinalli G, Sebag G, Zerah M, et al. Prognosis for mental function in Apert's syndrome. J Neurosurg. 1996; 85(1):66–72.