Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, primarily occurring in older adults, limited to the lungs, and associated with histological and/ or radiologic pattern of usual interstitial pneumonia. The objective of the study is to correlate severity of IPF and duration of symptoms with spirometry test/ forced vital capacity (FVC), high resolution computed tomography (HRCT) findings, smoking history, age and sex of 30 consecutive IPF patients in a tertiary care centre.
Methodology: The study was done on 30 consecutive patients of IPF, clinically and radiologically fulfilling the criteria for IPF.
Results: The study group showed significant male predominance of 80%. Subpleural, basal predominance, reticulations and honey-combing pattern on HRCT was seen in 100%, while traction bronchiectasis in 76.7% and minimal ground glassing in 33.3%. 23 out of 30 patients presented with symptom duration of 13-18 months, with mean duration of 16.4 months. 53.3% of the study population were smokers, 26.6% ex-smokers.
Conclusion: Clinicians should integrate clinical, radiological and/ or pathological data to support the diagnosis of IPF. Smokers are at particular risk for IPF and should be watched for at every visit. As IPF evolves over a period of time, early CT chest may not have all the radiological features supporting IPF. Close follow up is therefore suggested.
Keywords: Idiopathic pulmonary fibrosis; Forced vital capacity; High resolution computed tomography; Bronchiectasis
Full Text
Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, primarily occurring in older adults, limited to the lungs, and associated with histological and / or radiologic pattern of usual interstitial pneumonia [1]. IPF is a chronic progressive diffuse parenchymal lung disease of unknown origin with a heterogenous clinical pattern, with mortality rate exceeding that of many cancers [2-4]. Although the exact incidence and prevalence of the disease is not known, patients with this disease comprise about 15% of a pulmonary physician’s practice [5]. Mortality from cryptogenic fibrosing alveolitis continues to increase in many countries [6]. In India, this was earlier considered to be a rare disease. In 1979, Jindal et al. published their data on 61 cases of diffuse parenchymal lung disease (DPLD) seen over a period of five years [7]. However, the scenario is different now and the disease is no longer rare or uncommon.
Most have a gradual onset, often greater than 6 months of dyspnoea and/ or nonproductive cough. On high resolution computed tomography (HRCT), IPF is characterized by usual interstitial pneumonia (UIP) pattern of subpleural, basal distribution of reticular opacities associated with traction bronchiectasis. As IPF progresses, honey combing becomes prominent [2-4].
Currently, it is believed that epithelial injury and activation in fibroblast foci are crucial early events. Integrating the clinical, laboratory, radiological and/ or pathological data is the key to the diagnosis of IPF [8-10]. In the last decade, major advances in our understanding of the pathogenesis of IPF have led to a paradigm shift from a primarily inflammatory process evolving to fibrosis to a condition driven by aberrant wound healing following alveolar epithelial cell injury that results in scarring of the lung, architectural distortion, and irreversible loss of lung function [11]. Though pirfenidone and nintedanib, the two compounds with pleiotropic anti-fibrotic properties have been proven effective in reducing functional decline and disease progression in IPF, we still have a long way to go as both of these do not offer a cure for IPF and most patients continue to progress despite treatment [11]. The objective of the study is to correlate severity of IPF and duration of symptoms with spirometry test/ FVC, HRCT findings, smoking history, age and sex of 30 consecutive IPF patients in a tertiary care centre.
Materials and methods
A cross-sectional study was conducted at tertiary care centre (Krishna Institute of Medical Sciences Ltd.) Secunderabad over 6 months period on 30 patients. Selected patients were explained the purpose of the study and the need of co-operation was emphasized. All subjects participated in the study voluntarily. Written informed consent was obtained from all patients. Ethical and scientific clearance for the study was obtained from the Ethical Review Committee KIMS.
The patients were selected who were between age 40-70 years and who were clinically and radiologically fulfilling the criteria for the diagnosis of IPF and UIP pattern on CT chest. Patients who have the history of other interstitial lung disease, chronic obstructive pulmonary disease (COPD), asthma, pulmonary Koch’s, congestive cardiac failure (CCF), coronary artery disease (CAD), connective tissue disorders, drug toxicity, occupational disease and retro viral disease were excluded from the study.
The following investigations were done in all patients: 1) Spirometry test/ forced spirometry vital capacity, 2) HRCT scan chest. Spirometry test is performed using HELIOS 401 spirometer (RMS company), which records a volume-time curve, showing volume (litres) along the Y-axis and time (seconds) along the X-axis, and a flow-volume loop, which graphically depicts the rate of airflow on the Y-axis and the total volume inspired or expired on the X-axis.
FVC: The amount of air which can be forcibly exhaled from the lungs after taking the deepest breath possible, is measured by spirometry. Severity of FVC can be interpreted as mild: 70-79% predicted, moderate: 60-69% predicted, moderately severe: 50-59% predicted, severe: 35-49% predicted, very severe: <35% predicted.
HRCT: CT model being used at KIMS Hospital is Philips, Ingenuity CT. HRCT allows a detailed examination of the lung parenchyma by creating 1-2 mm thin slices of the chest. HRCT protocol followed by KIMS (Filter: lung window, Thickness: 1mm, Increment: 10nm, Window width: 1600, Window level: 600, Results of direction: Head to feet).
Statistical analysis: Master sheet of data was made with microsoft excel, graphs and charts were made with MS excel and word.
Results
After retrieving all the data and analyzing, the descriptive statistics of the study population was calculated. The total number of subjects in the study was 30. The total number of male and female are 24 and 6 respectively and the percentage wise distribution is given in figure 1.
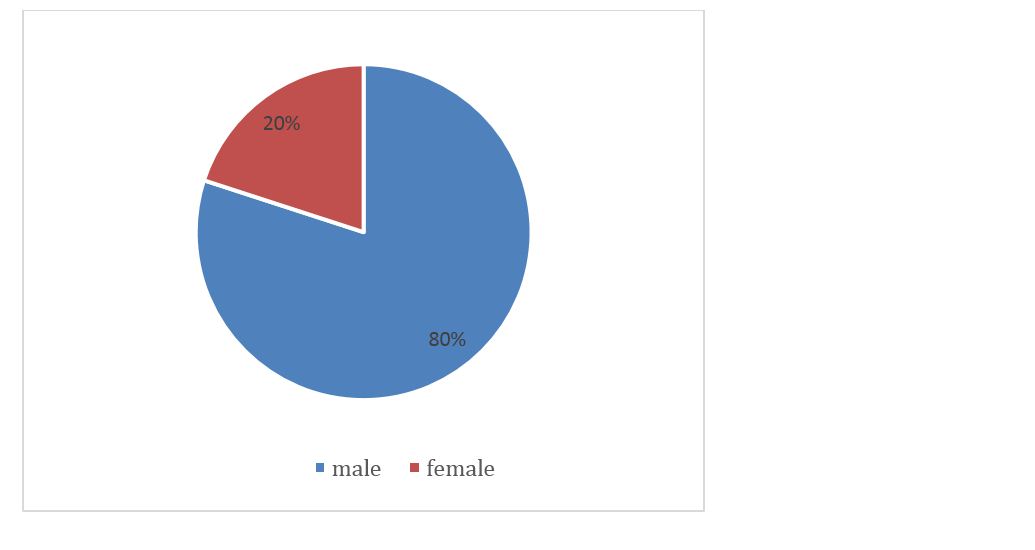
Figure 1: Gender distribution of patients.
The age group 66-70 recorded the highest number of patients with IPF in the study population. 51-55 recorded the least patients in the study population. The detailed information is given in figure 2.
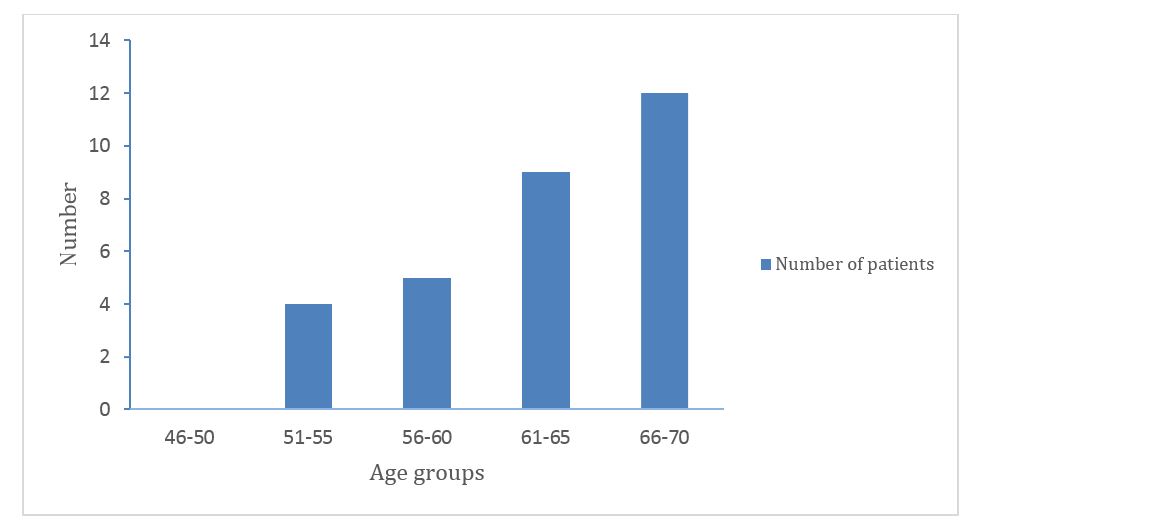
Figure 2: Age wise distribution of study population.
Out of 30 patients, 14 had experienced the symptoms for 13-14 months whereas 3 patients experienced the symptoms for more than 22 months. The detailed information is graphed in figure 3.
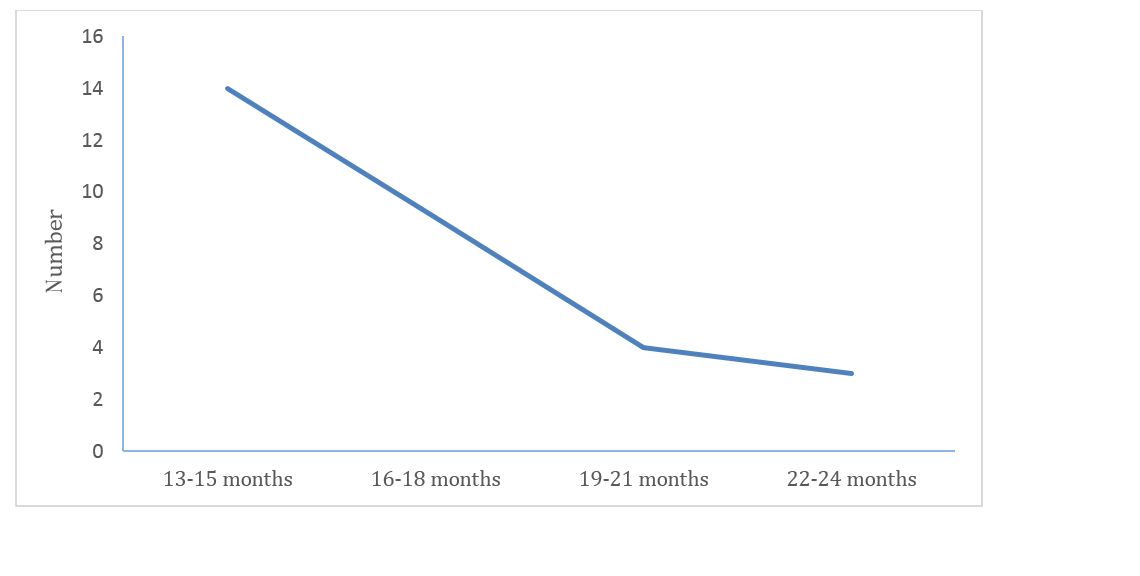
Figure 3: Representation of duration of symptoms in patients with IPF.
33.3% of patients had mild restriction where as 6.7% of patients had very severe restriction when FVC was measured. The detailed information is given in figure 4.
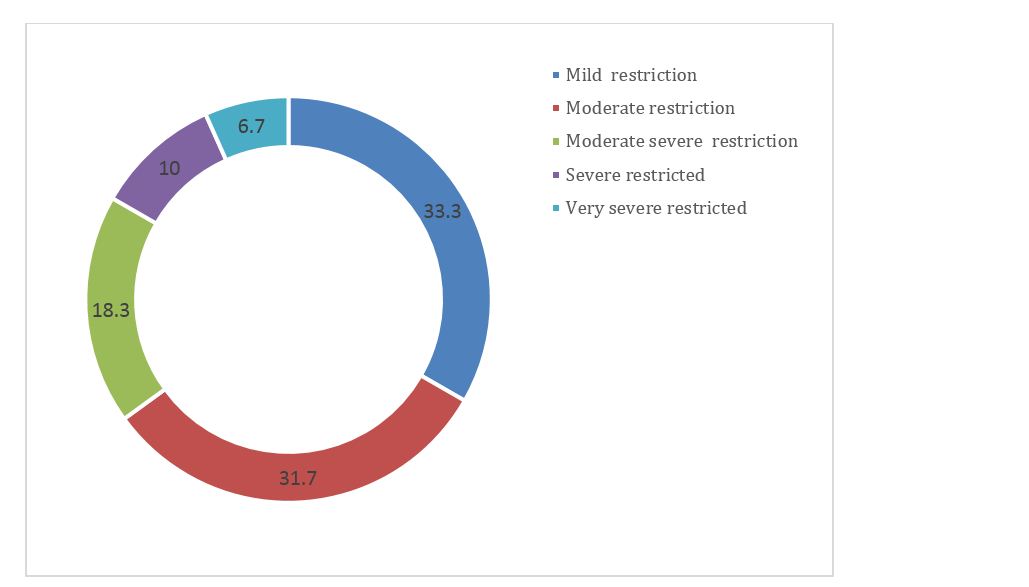
Figure 4: Spirometry grading of FVC.
Subpleural basal predominance, reticulations and honey combing pattern on HRCT was seen in 100%, while traction bronchiectasis in 76.7% and minimal ground glassing in 33.3%. Further information is shown in figure 5.
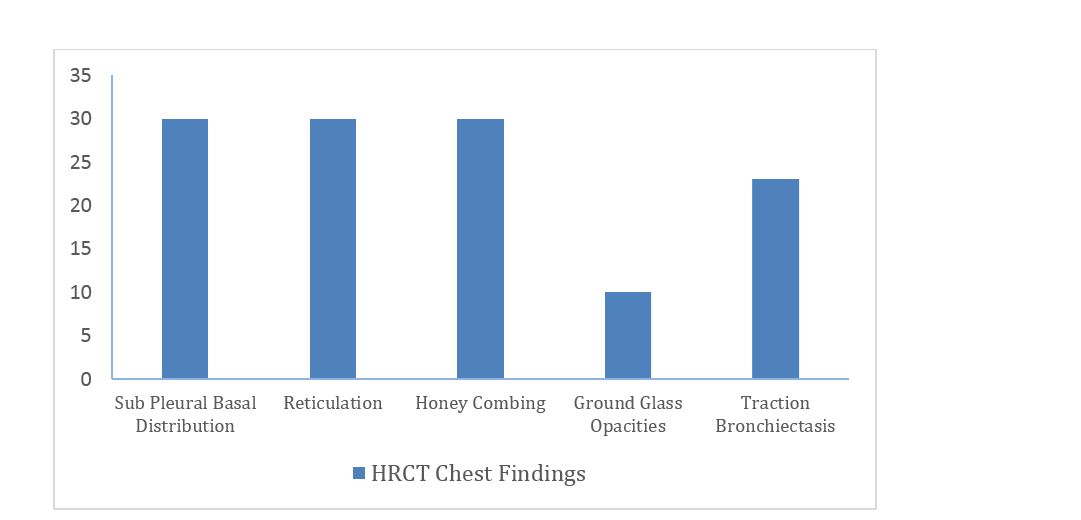
Figure 5: HRCT chest findings of IPF patients.
23 out of 30 patients presented with symptom duration of 13-18 months, with mean duration of 16.4m. 53.3% of the study population were smokers, 26.6% ex-smokers. Detailed information can be seen in figure 6.
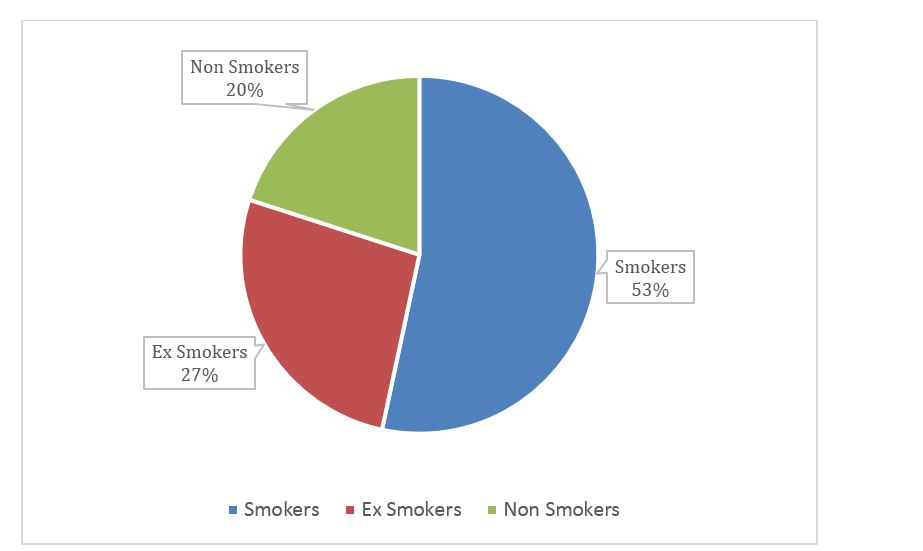
Figure 6: Graphical representation of smoking history in IPF patients.
Discussion
In this study, we included 30 subjects who have been clinically and radiologically diagnosed as IPF. Significant gender predominance is present (M > F) with male being 80% and females being 20% with (P value of 0.05) and disease occurred in seventh decade (70%) subpleural, basal reticulation and honey combing was seen in 100% in HRCT. The prevalence and incidence of IPF was higher in men (20.2 cases per 100,000 persons and 13.2 cases per 100,000/yr respectively) than women (10.7 per 100,000 persons and 7.4 per 100,000/yr, respectively). Both the prevalence and incidence of IPF increased dramatically with age [12].
Spirometry (FVC) and HRCT chest are the two noninvasive techniques which are useful in the diagnosis of IPF. In our study subpleural, basal predominance, reticulations and honey combing pattern on HRCT chest were present in all 30 patients (100%) followed by traction bronchiectasis (76.7%) and ground glassing (33.3%). In a prospective study conducted by Raniga S, et al. [13] in 10 IPF patients at a tertiary care government hospital of 7 women and 3 men, found that IPF is most commonly manifested by the combination of HRCT findings, which includes peripheral (100%) and lower lobe predominance (80%) of the distribution of the lesions, evidences of intralobular septal thickening (100%), honey combing (90%), traction bronchiectasis (90%) and parenchymal distortion (100%) and concluded that HRCT is accurate and superior in the conflict diagnosis of IPF.
In our study all patients had lung restriction in spirometry (p value = 0.05) but varied in the severity of restriction with predominance of mild restriction (FVC<80% predicted) in 10 patients closely followed by moderate restriction (FVC 60-90% predicted) in 9 patients and at the other end of the spectrum 3 patients with severe restrictions (i.e. FVC 35-49% predicted).
In a retrospective study conducted by Flaherty KR, et al. [14] found that for patients with usual interstitial pneumonia, change in FVC was the best physiological predictor of mortality (p = 0.05) and concluded that 6 months change in FVC gives additional information to baseline features for patients with idiopathic interstitial pneumonia. Similarly the patients in our study presented with symptoms duration 13 to 18 months as compared to 14 patients between 19 to 24 months. Mean duration of symptoms is of 16.4 months.
Smoking as a risk factor for IPF was also studied by Baumgarten, et al. [15] which stated that risk was significantly elevated for former smokers (OR = 1.9) and for smokers with 21 to 40 per years (OR = 2.3), correlating with our study we found 57% of subjects were smoker and 30% of subjects were ex-smokers. But the study period is short to draw any conclusion.
Conclusion
Spirometry showed restriction pattern in both genders. Mild and moderate restrictions of lung function (according to FVC) were more common as compared to sever restriction. Most patients presented with symptoms early in their disease course between 13 to 15 months and had a milder disease (according to FVC). All patients had subpleural basal predominance, reticular and honey combing findings followed by traction bronchiectasis in HRCT chest. Smokers and ex-smokers were more in number as compared to non-smokers. The disease is more severe in smokers and ex-smokers as compared to non-smokers. Most of IPF patients fall in age group between 60-70 years. Most of the females are in age groups of 51-55 years while males in age group between 66 to 77 years. Significant male predominance compared to females.
Acknowledgements
Acknowledgements are due to the Departments of Radiology & Imageology, and Pulmonology, Krishna Institute of Medical Sciences (KIMS), Secunderabad.
Conflict of interest
There was no conflict of interest and all the authors were in tone with the study.
References
1. Raghu G, Weycker D, Edelsberg J, Braadford wz, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am j Respir crit care med. 2006; 174(7):810–816.
2. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000; 161:646–664.
3. American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002; 165:277–304.
4. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3:322–329.
5. Coultas DB, Hughes MP. Accuracy of mortality data for interstitial lung diseases in New Mexico, USA. Thorax. 1996; 51(7):717–720.
6. Hubbard R, Johnston I, Coultas DB, Britton J. Mortality rates from cryptogenic fibrosing alveolitis in seven countries. Thorax. 1996; 51:711–716.
7. Jindal SK, Malik SK, Deodhar SD, Sharma BK. Fibrosing alveolitis: a report of 61 cases seen over the past five years. Ind J Chest Dis Allied Sci. 1979; 19:174–179.
8. Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: Impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med. 2000; 161(4 Pt 1):1172–1178.
9. King TE Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001; 164(7):1171–1181.
10. Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, et al. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006; 61(11):980–985.
11. Tzouvelekis A, Bonella F, Spagnolo P. Update on therapeutic management of idiopathic pulmonary fibrosis. Ther Clin Risk Manag. 2015; 11:359–370.
12. Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung disease. Am J Respir Crit Care Med. 1994; 150:967–972.
13.Raniga S, Sharma S, Arora A, Khalasi Y, Vora PA. “Utility of high resolution computed tomography (HRCT) in diagnosis and management of idiopathic pulmonary fibrosis”- a study of 10 cases. Indian J Radiol Imaging. 2006; 16:841–846.
14. Flaherty KR, Mumford JA, Murray S, Kazerooni EA, Gross BH, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Resp Crti Care Med. 2003;168 (5):543–548.
15. Baumgartner KB, Samet JM et al. “Cigratte smoking: a risk 2012 Apr 1; 53(3):296–302.