Full Text
Introduction
Most physicians will see diabetic emergencies on a regular basis, as they are common. However, nondiabetic endocrine emergencies are rarer and require a high index of suspicion. The basic managements for common endocrine emergencies are outlined in this article. The following topics are discussed:
- Myxedema coma
- Thyroid storm
- Acute adrenal insufficiency
- Pituitary apoplexy
- Pheochromocytoma crisis
- Acute hypercalcemia
- Acute hypocalcemia.
Myxedema coma
Myxedema coma (Figure 1) is a medical emergency with a high mortality rate [1]. It is defined as severe hypothyroidism with depressed mental status, hypothermia and reduced function of other organs. In the present day, this condition has become uncommon, due to early diagnosis, readily available tests and treatment at an affordable cost. Early diagnosis of myxedema coma is important. Previous history of thyroid surgery, administration of radioactive iodine and poor compliance to treatment with thyroid hormone will give a clue toward this condition. Apart from a depressed mental state and hypothermia, myxedema coma is associated with hyponatremia, hypotension and bradycardia. Myxedema means puffiness of the limbs and face which are due to abnormal deposits of mucin in the skin and other tissues. Despite the name myxedema coma, patients present with reduced consciousness, confusion, psychosis, etc. rather than a comatose state.
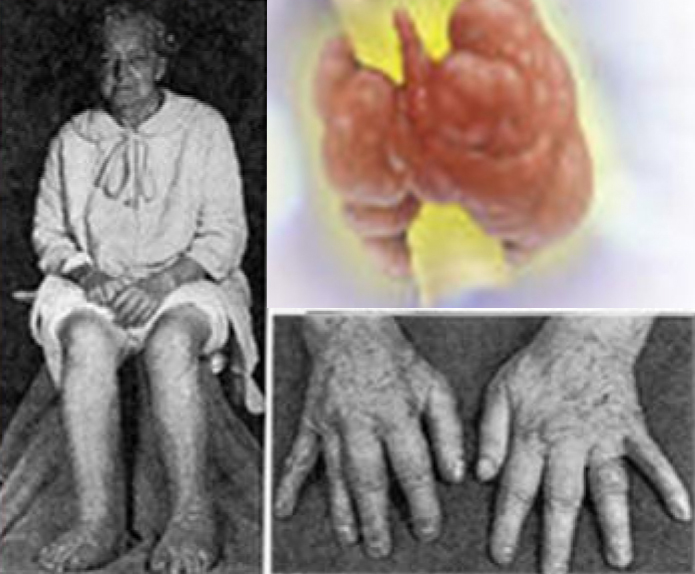
Figure 1: Myxedemic coma.
Hyponatremia is common and seen at least in half of the patients with this condition. In fact, any patient with a low serum sodium and normal volume status should be suspected of underactive thyroid. Hypothermia is another feature and has prognostic significance [2].
Other common features are hypoglycemia, bradycardia, hypotension and type 2 respiratory failure.
When clinical suspicion is high with the above mentioned features, treatment should be initiated quickly. However, blood should be drawn for thyroid stimulating hormone (TSH), free T4 and cortisol. If possible, a short synacthen test should be considered, as coexisting hypoadrenalism is a possibility, particularly so in secondary hypothyroidism due to pituitary cause. In spite of aggressive treatment with thyroid hormone replacement, supportive care and other measures, around a third may succumb to the illness. Treatment is preferably with a combination of T4 and T3. The dose of T4 is between 200 mcg and 400 mcg per day intravenously and the dose of T3 is 10 mcg three times a day intravenously. The young, obese and those with less likely cardiac risk should receive higher dose than their counterparts [3]. The usual practice is to give steroid cover in the form of hydrocortisone 100 mg every eighth hourly until hypoadrenalism is excluded. The crucial aspect of the management of this condition is however supportive treatment with correction of hyponatremia, hypothermia, bradycardia and hypotension [4]. Myxedema coma can occur as the culmination of severe, long-standing hypothyroidism or be precipitated by an acute event such as infection, myocardial infarction, cold exposure, or the administration of sedative drugs, especially opioids. (Figure 2).
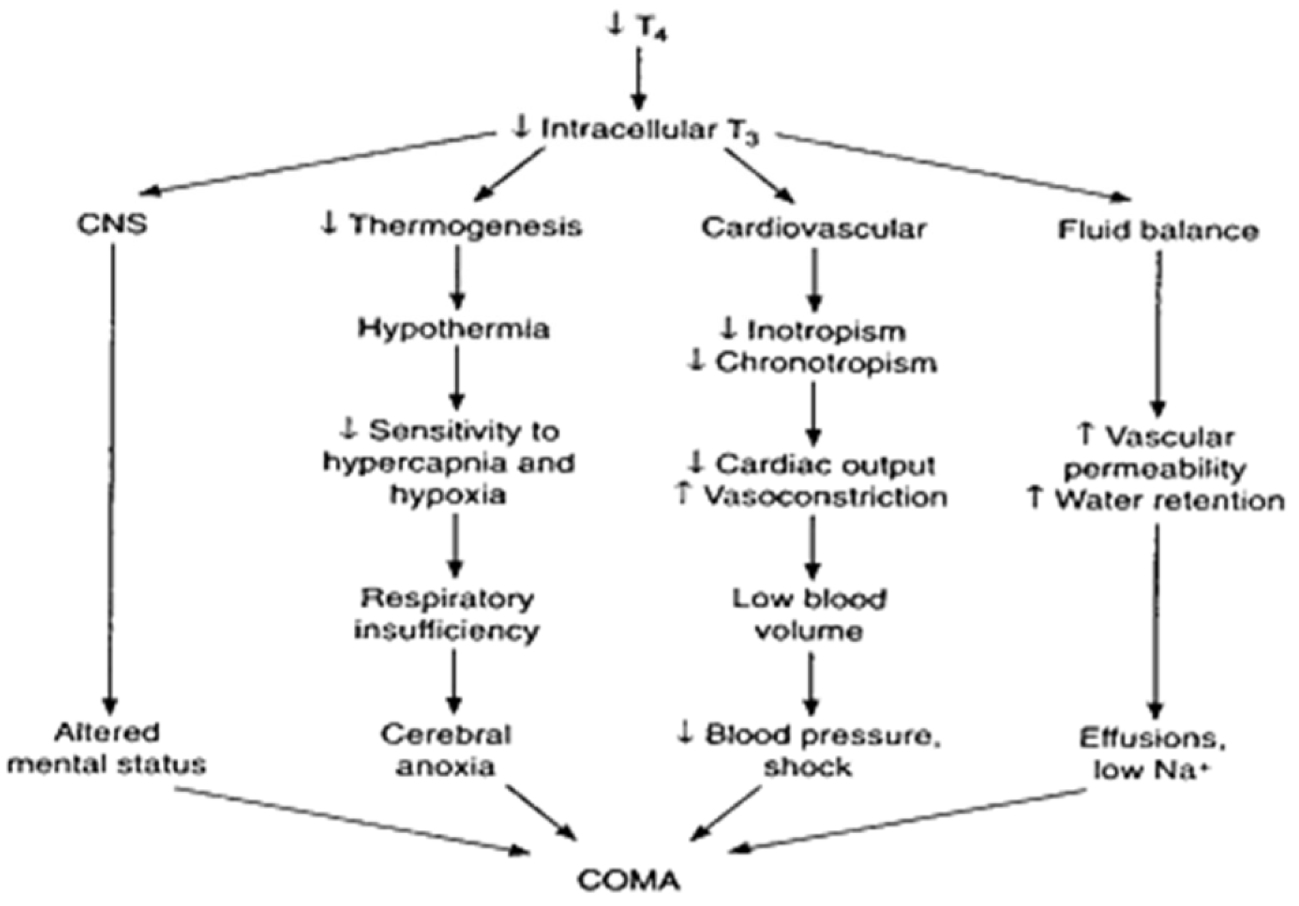
Figure 2: Pathogenesis of myxedema.
Thyroid storm
Thyroid storm is a life-threatening condition with severe symptoms and signs of thyrotoxicosis [5]. It is rare and the diagnosis is based upon clinical findings like tachycardia, hyperpyrexia, central nervous system dysfunction (agitation, delirium, psychosis, stupor or coma) and gastrointestinal symptoms (nausea, vomiting, abdominal pain). Physical examination may reveal a goiter, ophthalmopathy suggestive of Graves’ disease, lid lag, hand tremor and warm, moist palms. Thyroid function tests show hyperthyroidism (suppressed TSH, elevated free T4 and/or T3).
In addition to specific therapy directed against the thyroid, supportive therapy in an intensive care unit and recognition and treatment of any precipitating factors is essential, since the mortality is around one- third [6]. Treatment with a beta blocker, propranolol 80 mg orally every 6 hours to achieve adequate control of heart rate, carbimazole (20 mg every 6 hours) or propylthiouracil (200 mg every 4 hours), need to be administered. The latter drug is preferable for life-threatening thyrotoxicosis along with hydrocortisone 100 mg intravenously every 8 hours, until the patient is stable. In severe cases, 1 hour after a thionamide is given; administer iodine, Lugol’s solution 10 drops every 8 hours.
For patients with contraindications to thionamides, surgery is the treatment of choice [7]. Patients who are to undergo urgent surgery require preoperative treatment of thyrotoxicosis with beta blockers (if not contraindicated, propranolol 60–80 mg every 4–6 hours), glucocorticoids to inhibit peripheral conversion of T4 to T3 (e.g. dexamethasone, 1–2 mg every 6 hours) and iodine—supersaturated potassium iodide (SSKI), five drops (containing 20 drops/mL, 38 mg iodide/drop) orally every 6 hours or Lugol’s solution, 10 drops (containing 20 drops/mL, 8 mg iodine/drop) every 8 hours. Treatment is continued for 5–7 days with surgery performed on the sixth to eighth day. After the clinical manifestations of thyroid storm are improved, long-term therapy is either with drugs, radioactive iodine or surgery.
Key features of thyroid storm:
- Fever • Tachycardia
- Altered LOC
- Features of underlying hyperthyroidism
- Propylthiouracil (PTU) 1000mg loading and 1200mg/day tds or carbimazole.
- Weight loss, heat intolerance, tremors, anxiety, diarrhea, palpitations, sweating, CP, SOB
- Goiter, eye findings, pretibial myxedema
- Level of thyroid function is not discriminatory, but organ dysfunction is the criteria
Summary of management
- Supportive care
- Identification and treatment of the precipitating event
- Blocking the release and effects of thyroid hormone
- Propylthiouracil (PTU) 1000mg loading and 1200mg/day tds or carbimazole.
- Proproanolol IV 1mg every 10 to 15 min. until symptoms are controlled.
- Hydrocortisone 100mg IV 8 hourly. o Iodine acts by inhibiting hormone release but should not be given until 1 hour after PTU administration.
- Plasmapheresis/ plasma exchange/ hemodialysis.
Acute adrenal insufficiency
Adrenal crisis should be considered in any patient who presents in a state of shock even when there is no prior history of adrenal disease, particularly if there is coexisting unexplained severe hypoglycemia or hyponatremia [8]. The patient may present with abdominal pain and the crisis is usually precipitated by sepsis. Primary adrenal insufficiency is usually due to autoimmune conditions, tuberculosis, metastases or even drugs like antifungals. Secondary adrenal insufficiency is due to pituitary pathology where there is only glucocorticoid deficiency without any mineralocorticoid deficiency; hence, adrenal crisis is rare. Adrenal crisis can also occur in patients who have abruptly stopped their steroid medication [9]. The diagnosis is ideally made by a short adrenocorticotropin hormone (ACTH) stimulation test. However, there should be no reason to delay treatment in a sick patient. It is useful to take blood for electrolytes, cortisol and ACTH before initiating steroid treatment. Emergency treatment is by the administration of intravenous fluids, 0.9% saline and dextrose saline if there is hypoglycemia as well. Steroid treatment could be in the form of hydrocortisone 100 mg three times in a day until stable, when the treatment is switched to oral hydrocortisone at the dose of 10 mg in the morning, 5 mg at 12 noon and 6 pm daily, with patient education on sick day rule [10].
Key features of adrenal crisis
- Non specific
- Nausea, vomiting, abdominal pain
- Shock
- Distributive shock not responsive to fluids or pressors.
- Laboratory (variable)
- Hyponatremia, hyperkalemia, metabolic acidosis.
- known adrenal insufficiency
- Features of undiagnosed adrenal insufficiency
- Weakness, fatigue, weight, loss, anorexia, N/V, abdo pain, salt craving, hyperpigmentation.
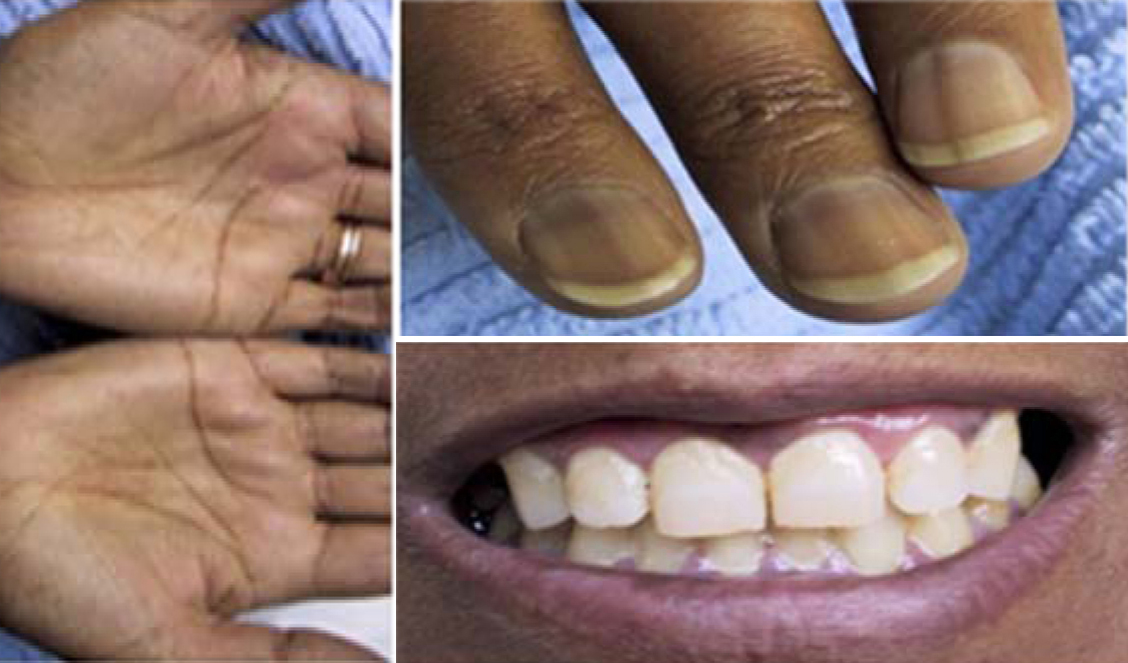
Figure 3: Hyperpigmentation, Generalized skin pigmentation (in a caucasion patient) but especially the deposition in the palmer skin creases, nails and gums. She was treated many years ago for pulmonary TB.
Management of Adrenal Crisis
- Corticosteroid replacement
- Dexamethasone 4mg iv q6hr is the drug of choice (Doesn’t affect ACTH stim test)
- Hydrocortisone 100mg iv is an option
- Mineralocortocoid not required in acute phase
- Other
- Correct electrolytes, fluid resuscitation(2-3L)
- Glucose for hypoglycemia
Pituitary apoplexy
Sudden hemorrhage into the pituitary gland is called pituitary apoplexy. The clinical presentation ranges from relatively mild symptoms to adrenal crisis, coma or even sudden death. The usual triads for apoplexy are sudden severe headache, diplopia and features of hypopituitarism [11]. Hemorrhage often occurs into a pituitary adenoma, resulting in hypopituitarism, but cortisol deficiency is the most serious amongst all hormone deficiencies because it can cause life-threatening hypotension [12]. Although surgery is the treatment of choice, conservative approach can also be effective. Hydrocortisone replacement should be promptly initiated as in adrenal crisis [13].
Hemorrihage into the pituitary gland is callced pituitary apoplexy. In its most dramatic presentation, apoplexy causes the sudden onset of excruciating headache, diplopia due to pressure on the oculomotor nerves, and hypopituitarism (Figure 4).
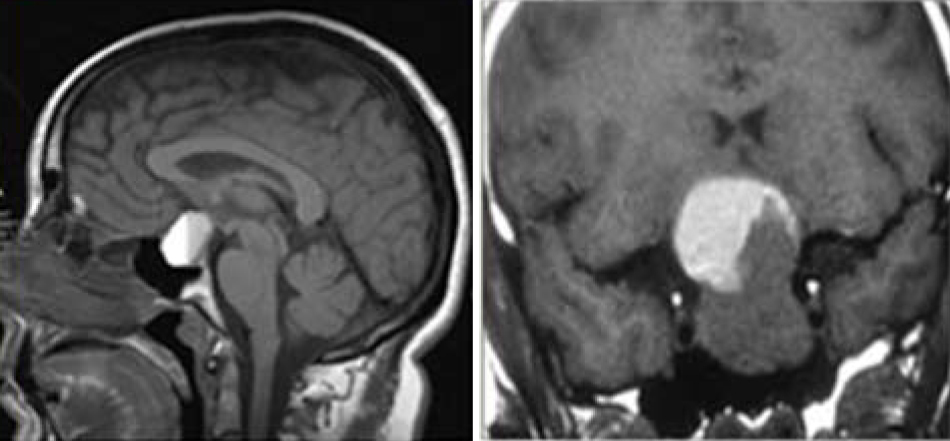
Figure 4: MRI- Pituitary hemorrhage
Management of pituitary apoplexia
Hormonal: Dexamethasone 4mg bd (glucocorticoid support and relief of cerebral edema) initially and maintance of hydrocortisone 10mg/5mg/5mg a day long term.
Neuro surgical: Transphenoidal pituitary decompression. After the acute episode the patient must be evaluated for multiple pituitary deficiencies.
Pheochromocytoma crisis
Although uncommon, the classic triad of symptoms in pheochromocytoma consists of episodic headache, sweating and tachycardia. Sustained or paroxysmal hypertension is the most common sign; however, around 10% may have normal blood Pressure [14]. Measuring the fractionated metanephrines in the plasma, is a first-line test when there is a high index of suspicion for pheochromocytoma and 24-hour urinary fractionated catecholamines and metanephrines for low index of suspicion. Once biochemical confirmation is made, computed tomography (CT) or magnetic resonance imaging (MRI) adrenals (Figure 5) should be performed and MIBG (123-I-metaiodobenzylguanidine) in those who do not have adrenal tumors [15].
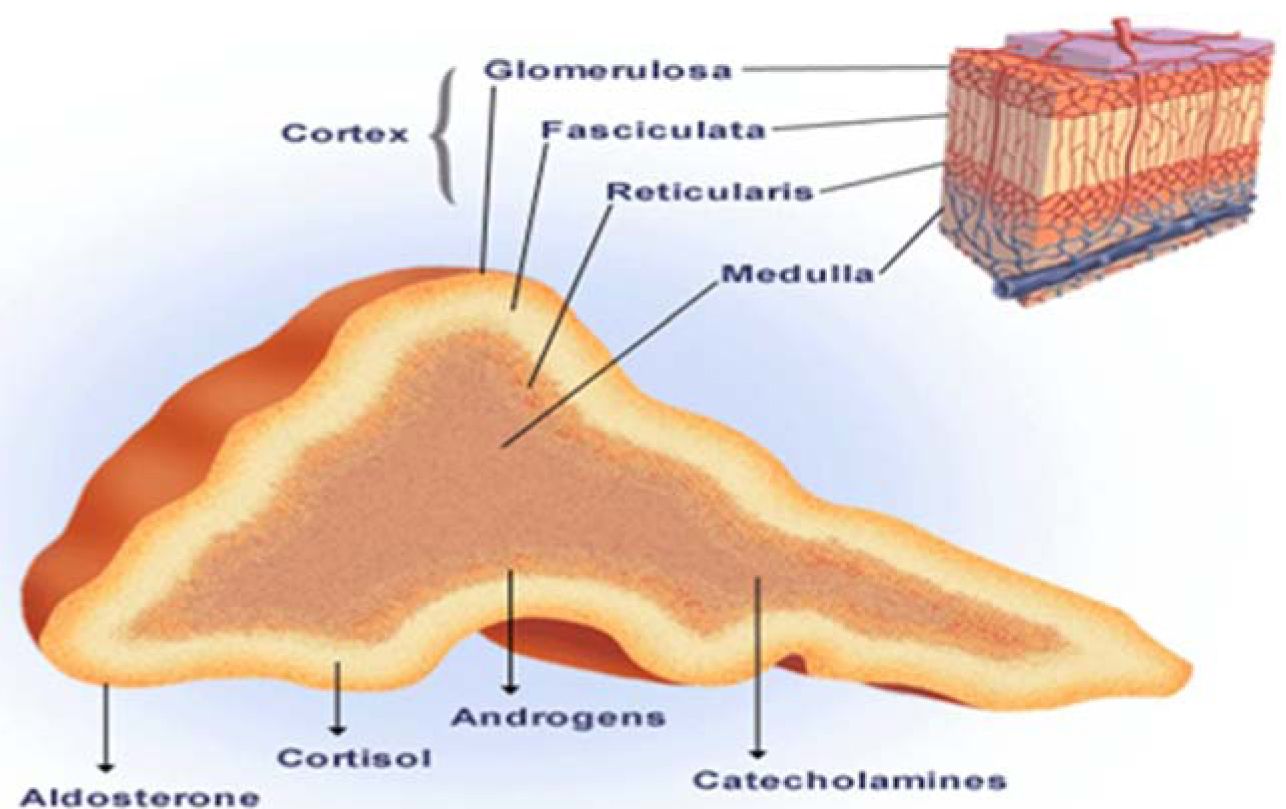
Figure 5: Adrenal physiology
The term pheochromocytoma crisis is used when patients present with potentially life-threatening hemodynamic disturbances either in the form of hypertensive crisis or cardiogenic (or adrenergic) shock. Mortality is around 85%. Treatment of resistant hypertension is with intravenous infusion of nitroprusside (arteriolar and venous dilator), at a dose of 0.25–0.5 μg/kg per minutes; maximum dose: 8–10 μg/ kg per minutes. Its duration of action is less than 5 minutes. Thus, hypotension can be easily reversed by temporarily discontinuing the infusion. The aim is to reduce diastolic blood pressure to around 100 mm Hg and systolic should be dropped by not more than 25%. However, the use of nitropursside should be brief, due to possible cyanide toxicity. Oral choices are nifedipine or labetalol. Once the patient is stable, oral phenoxybenzamine 10 mg twice daily is initiated and the dose titrated as per the blood pressure. Beta blockers like propranolol can be added later to optimize the patient for surgery [16].
Clinical features and diagnosis: The classic triad of symptoms in patients with a pheochromocytoma consists of episodic head ache, sweating, and tachycardia. History of poorly controlled hypertension or accelerated hypertension. Attacks build up over a few minutes and fade gradually over 15 minutes or can be more sustained (60 min). Diagnosis: 24 urine collection for free catecholamines and metanephrines (Figure 6).
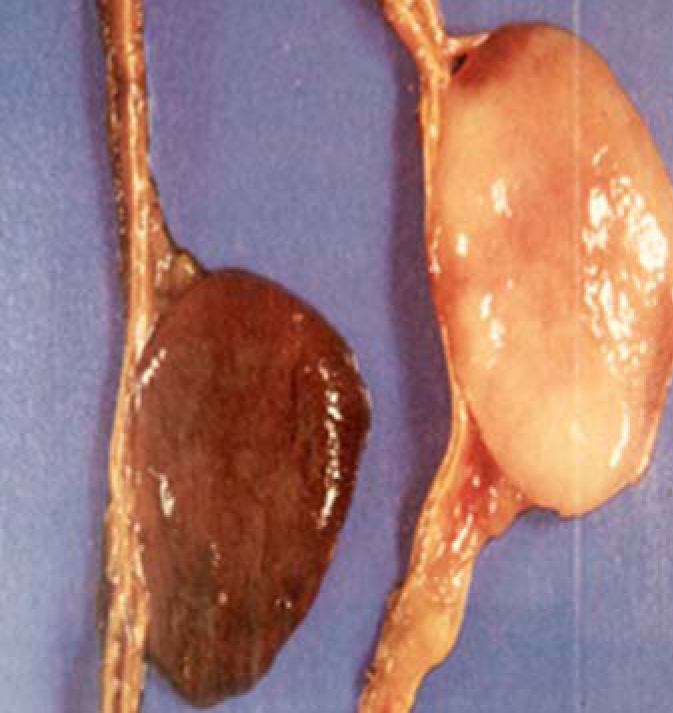
Figure 6: Diagnosis, CT/MRI Adrenals, MIBG Scan.
Treatment: Surgery after adequate blockade, α-antagonists: Phenoxybenzamine, Prazocin, Doxazosin, Non selective β –antagonists should precede β-antagonist treatment with 48h. Be aware of postural hypotension.
Acute hypercalcemia
Primary hyperparathyroidism [parathyroid hormone (PTH) mediated] and malignancy account for over 90% of cases. The latter is usually more severe and symptomatic while the former is long standing. Other causes are tertiary hyperparathyroidism due to end stage renal disease, as part of multiple endocrine neoplasias or due to familial hypocalciuric hypercalcemia [17].
The list for non-PTH mediated hypercalcemia is much more elaborate, but by far the most common cause is due to osteolytic bone metastases and related to PTHrp (PTH-related peptide), or due to increased calcitriol via extra renal conversion. Other causes of non- PTH mediated hypercalcemia include increased vitamin D levels due to over replacement or due to chronic granulomatous diseases. Drugs like thiazide diuretics, lithium, excessive vitamin A and conditions like hyperthyroidism, acromegaly and pheochromocytoma may also contribute.
The goal of treating hypercalcemia is twofold, one to lower hypercalcemia and the other is to treat the underlying cause. If facilities to measure ionized calcium are available, it is preferable, especially when abnormalities in albumin levels or conditions like myeloma are suspected. Hypercalcemia, less than 12 mg (3 mmol/L), in asymptomatic patients may not warrant immediate treatment. The initial treatment however is with 0.9% saline to correct dehydration and with furosemide in people who are at risk of fluid overload. Calcitonin at a dose of 4 IU (international units)/ kg subcutaneous/ intramuscular, twice daily, could be used (when available) in severe symptomatic hypercalcemia for the first 48 hours due to their immediate effects, after which tachyphylaxis may develop. Bisphosphonates are the most preferred agents for treating hypercalcemia and they work by inhibiting osteoclastic activity. The two popular bisphosphonates are pamidronate (30 mg, 60 mg or 90 mg intravenously over 2 hours) and zoledronic acid (4 mg IV over 15 minutes). Ibandronate is another agent which has similar efficacy as pamidronate [18].
Glucocorticoids will usually reduce serum calcium concentrations within few days by decreasing calcitriol production by the activated mononuclear cells in the lung and lymph nodes in chronic granulomatous conditions like sarcoid [19]. Calcimimetic agents like cinacalcet reduce the serum calcium concentration in patients with severe hypercalcemia and is useful in hemodialysis patients.
Common causes
- Endocrine:
- Hyperparathyroidism
- MEN
- PTHrp by Solid tumors
- Neoplastic:
- Ca with bone metastases
- Myeloma
- Granulomatous:
Clinical features: history of polyuria and polydipsia, Dehydration, Bone repair, confusion, Anorexia, Constipation.
Hypercalcemia management
- Saline rehydration
- Loop diuretics
- Pamidronate(IV biophosphonate) inhibits bone resorption and has a prolonged effect on calcium concentration
- Dialysis if pt has renal failure
- Calcitonin rarely used due to modest effect and rapid onset of tachyphylaxis (rapid decrease in response to the drug over a short period of time).
Acute hypocalcemia
Hypocalcemia is usually due to low PTH, secondary to thyroid, parathyroid or neck surgery, autoimmune conditions, underlying renal disease and more commonly due to vitamin D deficiency [20].
Sometimes, it is due to low magnesium levels as a result of diuretics, chronic diarrhea or malabsorption. The diagnostic approach of hypocalcemia is by first establishing low serum calcium and also measuring ionized calcium levels. Other investigations are, estimating PTH, vitamin D, phosphate levels, renal function and magnesium levels [21]. Treatment is based on the severity of symptoms and the level of hypocalcemia. Intravenous calcium replacement may be needed if calcium levels are below 7.5 mg/dL. Most patients with hypoparathyroidism would need lifelong treatment. The dose of oral calcium is 1 g of elemental calcium daily in divided doses. Calcitriol is administered at a dose of 0.25 mcg twice daily, with weekly dose increments, maintain serum calcium at the lower limit of normal [22].
Vitamin D deficiency is typically treated with 50,000 IU of vitamin D2 or D3 weekly for 6–8 weeks. In patients with hypocalcemia with magnesium deficiency, hypomagnesemia should be corrected first.
Causes of acute hypocalcaemia (Figure 7) and Biochemical workup Causes of acute hypocalcaemia Biochemical workup.
|
Causes of acute hypocalcaemia
|
Biochemical workup
|
|
Hypothyroidism
- Destruction of parathyroids
- Acute hypomagnesaemia
|
S total Ca++; albumin; ionized ca++; S PO4, S Mg++
|
|
Reduced 1,25(OH)vitamin D
|
plasma PTH
|
|
Increased uptake of Ca
- Osteoblastic metastases
- Hungry bone syndrome
|
25(OH)D3 and 1, 25(OH)D3; S amylase and lipase
|
|
Complexing of Ca
|
|
Figure 7: Acute hypocalcaemia.
Therapy
- IV calcium for acute, severe disease with continuous ECG monitoring
- Oral calcium for chronic disease with vitamin D.
- Thiazide diuretics to decrease amount of calcuria
- Goal calcium level is 8.5mg/dl and urinary calcium level is less than 300mg/24h.
Conclusions
Endocrine emergencies are relatively uncommon, yet they are important disorders which physicians encounter in the emergency department. Although many endocrine emergencies occur in patients with underlying endocrine pathology, it is important not to miss the diagnosis in de novo presentations. Measures such as aggressive supportive care, replacement of steroids and replacement of the appropriate endocrine hormones have been shown to decrease the morbidity and mortality associated with these diseases. In short, although endocrine emergencies are uncommon, the clinical diagnosis should never be missed, as delay can lead to worsened morbidity and mortality.
Conflict of interest
Author declares no conflict of interest.
References
1. Forester CF. Coma in myxedema: report of a case and review of the world literature. Arch Intern Med 1963, 111:734-743.
2. Yamamoto T, Fukuyama J, Fujiyoshi, A. Factors associated with mortality of myxedema coma: report of eight cases and literature survey. Thyroid 1999, 9: 1167-1174.
3. Arlot S, Debussche X, Lalau JD, et al. Myxoedema coma: response of thyroid hormones with oral and intravenous high-dose L- thyroxine treatment. Intensive Care Med. 1991, 17:16-28.
4. Hylander B, Rosenqvist U. Treatment of myxoedema coma-factors associated with fatal outcome. Acta Endocrinol (Copenh) 1985, 108: 65-71.
5. Nicoloff JT. Thyroid storm and myxedema coma. Med Clin North Am. 1985, 69:1005-1017.
6. Yeung SC, Go R, Balasubramanyam A. Rectal administration of iodide and propylthiouracil in the treatment of thyroid storm. Thyroid 1995, 5:403-415.
7. Gilliland PF. Endocrine emergencies. Adrenal crisis, myxedema coma, and thyroid storm. Postgrad Med 1983, 74: 215-220.
8. Bouillon R. Acute adrenal insufficiency. Endocrinol Metab Clin North Am. 2006, 35: 767-775.
9. Zwaan CM, Odink RJ, Delemarre-van de Waal HA, et al. Acute adrenal insufficiency after discontinuation of inhaled corticosteroid therapy. Lancet 1992, 340: 1289-1290.
10. Gardner DG, Shoback D. Acute Adrenal Insufficiency. In: Greenspan’s Basic and Clinical Endocrinology, 9th edition. McGraw-Hill, 2011 p. 238.
11. Randeva HS, Schoebel J, Byrne J, et al. Classical pituitary apoplexy: clinical features, management and outcome. Clin Endocrinol (Oxf ) 1999, 51:181-188.
12. Semple PL, Webb MK, de Villiers JC, et al. Pituitary apoplexy. Neurosurgery. 2005, 56: 65-72.
13. Ayuk J, McGregor EJ, Mitchell RD, et al. Acute management of pituitary apoplexy–surgery or conservative management? Clin Endocrinol (Oxf ) 2004, 61: 747-752.
14. Kobal SL, Paran E, Jamali A, et al. Pheochromocytoma: cyclic attacks of hypertension alternating with hypotension. Nat Clin Pract Cardiovasc Med 2008, 5:53-57.
15. Hebert CJ, Vidt DG. Hypertensive crises. Prim Care 2008, 35: 475-487.
16. Newell KA, Prinz RA, Pickleman J, et al. Pheochromocytoma multisystem crisis: a surgical emergency. Arch Surg 1988, 123: 956-959.
17. Bilezikian JP. Management of acute hypercalcemia. N Engl J Med 1992, 326: 1196-1203.
18. Edelson GW, Kleerekoper M. Hypercalcemic crisis. Med Clin North Am. 1995, 79: 79-92.
19. Inzucchi SE. Management of hypercalcemia. Diagnostic workup, therapeutic options for hyperparathyroidism and other common causes. Postgrad Med 2004, 115: 27-36.
20. Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008, 336: 1298-1302.
21. Shane, E. Hypocalcemia: pathogenesis, differential diagnosis, and management. In: Favus MJ (Ed). Primer on the metabolic bone diseases and disorders of mineral metabolism, 4th edition. Philadelphia: Lippincott Williams & Wilkins; 1999, p. 223-226.
22. Ariyan CE, Sosa JA. Assessment and management of patients with abnormal calcium. Crit Care Med 2004, 32: S146-154.