Full Text
Mixed gonadal dysgenesis (45X/46XYmosaicism and its variants) is a rare form of sex chromosome DSD (disorders of sex development), that probably arises through anaphase lag during mitosis in the zygote. Though the classic form is 45X/46XY, other variants in this category include mosaics of 45X/47XXY, 45X/47XXY. The exact prevalence of mixed gonadal dysgenesis is not known, as only those individuals with the most severe forms present to the clinician.
Gynaecomastia is enlargement of glandular tissue in retro areolar area of male breast, and is due to increased ratio of estrogens to testosterone. Etiology of gynaecomastia could be physiological (occurring normally during infancy, puberty, and older age), or pathological (disorders such as androgen deficiency, testicular tumors, hyperthyroidism, and chronic kidney disease) or pharmacological (drugs such as anti androgens, estrogenic preparations, spiranolactone, HAART (highly active antiretroviral therapy) therapy etc.). However 25% cases of gynaecomastia are idiopathic in nature. True gynecomastia should be differentiated from pseudogynecomastia, which refers to fat deposition without glandular proliferation.
Here we discuss a case of gynaecomastia in a 16-year-old patient of mixed gonadal dysgenesis, and the subsequent management issues of such patients.
Case report
A 16-year-old male patient (Figure 1) was referred to the department of endocrinology, with progressive bilateral breast enlargement of 1 year duration with no galactorrhoea. He denied any history of recurrent jaundice or usage of any drugs. On examination, his height was 164cm, weight was 62kg with a body mass index (BMI) of 23kg /m², arm span was 175cm and upper segment to lower segment ratio (US/LS) of 0.85. His pubertal staging was G³A¹P³ Tanner gonads stage 3, axillary hair stage 1, pubic hair stage 3, stretched penile length was 8cm and testicular volume was 8ml bilaterally, with a smooth surface. Gynecomastia was bilaterally 8cm in diameter, without any spontaneous or expressive galactorrhea. His vitals were normal with a blood pressure of 110/80mmHg, systemic examination was normal.
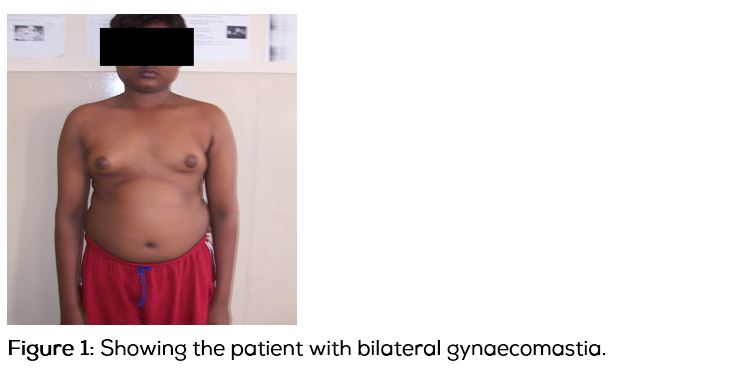
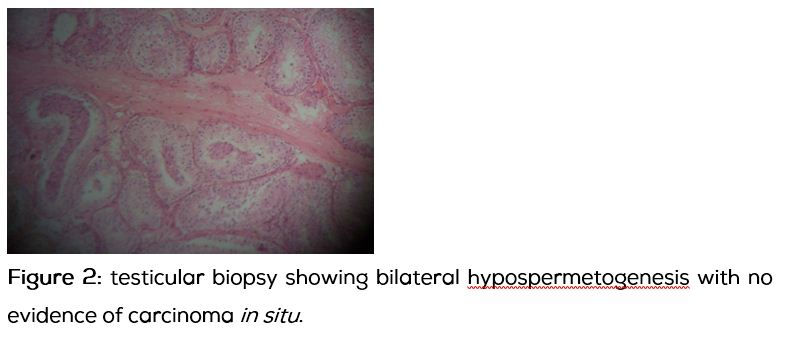
Investigations: Routine biochemistry was normal, with normal renal and liver functions. Testosterone was 1.7ng/ml, Estradiol – 82.9pg/ml, LH -2.6miu/ml, FSH 9.2miu /ml, βhCG 5.05miu/ml, Karyotype showed 45, X/46, XY mosaicism with distribution of 50% each. Ultrasound of the testis showed a normal homogenous appearance. Testicular biopsy revealed features suggestive of bilateral hypospermetogenesis observed in 20-30% of seminiferous tubules. There was no evidence of carcinoma in situ (CIS) (Figure 2). We diagnosed this as a case of 45, X/46, XY mosaicism with hypogonadism. Patient underwent reduction mammoplasty and he was started on androgen replacement and annual follow up with USG of testis has been planned.
Discussion
45,X/46,XY Mosaicism (Mixed gonadal dysgenesis) probably arises mainly through anaphase lag and is frequently associated with structurally abnormal Y-chromosome. Inter chromosomal rearrangements with loss of the structurally abnormal Y may be a common mechanism for the production of 45,X /46,XY mosaicism .
A highly diverse phenotype is encountered in this form of mosaicicm, ranging from phenotypic female to individual with ambiguous external genitalia to phenotypic males. In the review by Zah and co-workers of 60 patients with 45, X/46,XY Mosaicism, two thirds were reared as females. Turner’s stigmata and short stature are found in about half of the reported cases, in keeping with the chromosomal mosaicism containing an XO cell line. The presence of a Y-chromosome indicates the likely presence of testicular tissue and hence predicts the occurrence of virilization at puberty as well as high risk of gonadal tumours [1, 2]. The gonads in female phenotype vary from bilateral streaks to bilateral dysgenetic testes. Mixed gonadal dysgenesis is common in this mosaicism but is not specific for this, can also occur with 46,XY karyotype (eg. in familial 46XY dysgenesis) [3]. The development of male ducts and involution of the mullerian structures parallel the degree of testicular development on each side. Breast development at or after the age of puberty occurs in about one fourth of cases and is usually associated with a gonadal neoplasm.
The patients with 45, X/46, XY mosaicism are at a high risk for development of gonadal tumours. Carcinoma in situ (CIS) is thought to be a premalignant lesion leading to germ cell tumours [4, 5]. Gonadoblastoma is the neoplasm most often found, and it can lead to malignant germinoma. The risk of this tumor is 15-20% and it increases with age. The TSPY (testes specific protein Y encoded) gene is expressed in the foetal and adult testes and with high intensity in CIS of testes, gonadoblastoma and seminoma. Lau et al. reviewed evidence in support of aberrant expression of TSPY in dysgenetic testes acting as proto-oncogene [6].
Muller et al. recommended ultrasonography of the gonads and biopsy of the retained testis at the start of puberty. If biopsy and ultrasonography show lack of evidence of CIS, they suggest an annual ultrsonographic examination and second biopsy at 20 years of age [4]. The absence of CIS at age 20 suggests that the risk of gonadal germ cell tumours is minimal.
Treatment and follow up
The diagnosis is established by the demonstration of 45, X/46, XY mosaicism in blood, skin or gonadal tissue. As pointed out in the discussion, 45, X/46, XY mosaicism can present with wide variation in extent of sexual differentiation and the decision as to the sex of the rearing should be based upon the potential for normal function of the external genitalia. In the patients assigned a female gender role, the gonads should be removed, external genitalia should be repaired and oestrogen therapy should be initiated at the age of normal puberty. In patients assigned a male gender, all gonadal tissue except that which appears histologically normal and is in the scrotum should be removed, and prosthetic testes should be placed in the reconstructed scrotal sac, if appropriate. Even in the absence of evidence of testicular dysgenesis, close follow up is indicated, including a testicular biopsy at puberty and at age 20 to ascertain malignant potential [4, 5]. The need for androgen replacement therapy at adolescence depends on the capacity of the testes to secrete testosterone.
Conclusion
This is a patient of 16-year-old with normal male phenotype who presented with gynaecomastia of pubertal onset, having eunuchoid body habitus, with karyotype being 45, X /46, XY mosaicism. Ultrasonography of his testis was normal no evidence of carcinoma insitu (CIS) on gonadal biopsy. He was started on androgen replacement and annual follow up with USG of testis has been planned.
Conflict of Interest
The authors declare no conflict of interest.
References
1. Josso N, Nezelof C, Picon R, de Grouchy J, Dray F, et al. Gonadoblastoma in gonadal dysgenesis: a report of two cases with 46,XY-45,X mosaicism. J Pediatr. 1969; 74(3):425–437.
2. Rajfer J, Walsh PC. Mixed gonadal dysgenesis: dysgenetic male pseudohermaphroditism. In: Josso N, ed. The Intersex Child: Pediatric and Adolescent Endocrinology. Basel, Switzerland: S. Karger; 1981:105–115.
3. Canto P, de la Chesnaye E, López M, Cervantes A, Chávez B, et al. A mutation in the 5’ non high mobility group box region of the SRY gene in patients with Turner’s syndrome and Y mosaicism. J Clin Endocrinol Metab. 2000; 85(5):1908–1911.
4. Müller J, Skakkebaek NE, Ritzén M, Plöen L, Petersen KE. Carcinoma in situ of the testis in children with 45,X/46,XY gonadal dysgenesis. J Pediatr. 1985; 106(3):431–436.
5. Muller J, Ritzen EM, Ivarrsson SA, Rajpert-De Meyts E, NorjavaaraE, et al. Management of males with 45,X/46,XY Gonadal dysgenesis. Horm Res. 1999; 52(1):11–14.
6. Lau YF. Gonadoblastoma, testicular and prostate cancers, and the TSPY gene. Am J Hum Genet. 1999; 64(4):921–927.