Case Report
2016
December
Volume : 4
Issue : 4
Papillary tumor of pineal region: Case reports
Kashif M Mohiuddin, Sailaja M, Satish Rao I and Manas Panigrahi
Pdf Page Numbers :- 169-173
1Department of Pathology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India
2Department of Neurosurgery, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India
*Corresponding author: Dr. Kashif M Mohiuddin, Department of Pathology, Krishna Institute of Medical Sciences, Minister Road, Secunderabad-500003, Telangana, India. Tel.: 9848969543; Email: kashifhazim@gmail.com
Received 11 July 2016; Revised 22 August 2016; Accepted 31 August 2016; Published 07 September 2016
Citation: Mohiuddin KM, Sailaja M, Satishrao I and Manas P. Papillary tumor of pineal region: Case reports. J Med Sci Res. 2016; 4(4):169-173. DOI: http://dx.doi.org/10.17727/JMSR.2016/4-037
Copyright: © 2016 Mohiuddin KM, et al. Published by KIMS Foundation and Research Center. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: The papillary tumor of the pineal region (PTPR) was a new distinct entity described in 2003 WHO classification and later included in the subsequent 2007 and 2016 WHO classification of Central Nervous System tumours. They are commonly associated with obstructive hydrocephalus and are difficult to distinguish from other clinical entities, more particularly pineocytoma. Due to the limited number of reported cases, treatment strategies are not well established.
Case report: We report two cases of PTPR who presented with headache impaired gaze and diplopia. In first case MRI showed a cystic mass in the third ventricle and pineal region whereas the tumor had displaced the tectal plate in second case. The initial squash suggested a neoplastic lesion of probable glial origin favoring ependymoma. On microscopy, the tumor displayed papillary architecture with perivascular pseudo rosettes displaying multi layering of cuboidal to columnar cell with vesicular nucleus. Based on histological features the differential diagnostic conditions considered were choroid plexus tumor, papillary ependymoma, choroid plexus carcinoma, PTPR, and metastatic carcinoma. PTPR was confirmed on immunohistochemistry workup. Both the patients had recurrence, after one year and six months respectively. Conclusion: PTPR is an important differential diagnosis to be considered in cases where the tumoris arising in the vicinity of pineal region showing papillary architecture.
Keywords: pineal region papillary tumor; PTPR; pineal region; subcommissural organ
Full Text
Introduction
Tumors of the pineal region represent less than 1% of all intracranial tumors. Tumors arising in the vicinity of pineal gland may display prominent papillary features and can lead to diagnostic dilemma. Differential diagnoses include a variety of tumors having papillary configuration like papillary tumours of the pineal region (PTPR), pineal parenchymal tumours with papillary features, papillary ependymoma, papillary meningioma, choroid plexus papillomas and papillary metastatic deposits [1, 2]. Following are 2 case reports of tumors arising in vicinity of pineal gland and which had papillary configuration on histological examination.
Case 1
A 48-years-old female presented with impaired gaze and headache since two months. Magnetic resonance imaging (MRI) showed an enhancing solid cystic mass in the posterior third ventricle and pineal region (Figure 1a) was suggestive of glioma. Excision was done through Combined supra an infratentorial approach. On microscopy the tumor showed papillary architecture with central fibrovascular core containing hyalinized vessels. The papillae were lined by single to multilayers of columnar cells with moderate amount of granular, eosinophilic cytoplasm, round to oval hyperchromatic nuclei with finely granular chromatin and conspicuous nucleolus (Figure 1b, c). Cellular dyscohesion was evident. Areas of necrosis were also seen but with no significant mitotic activity. Differential diagnoses considered were papillary tumor of pineal region, choroid plexus tumor and papillary ependymoma. On immunohistochemistry (IHC) the tumor was diffusely positive for pancytokeratin (Figure 1e), focally positive for glial fibrillary acidic protein (GFAP) (Figure 1d) and epithelial membrane antigen (EMA) negative. IHC profile was consistent with papillary tumor of pineal region. The patient had recurrence after one year.
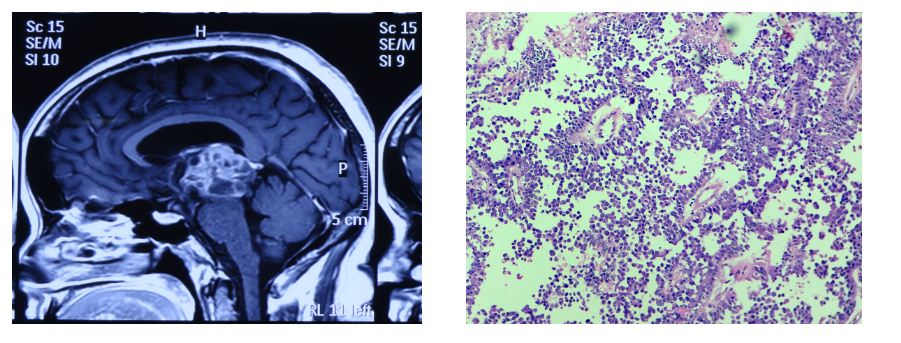
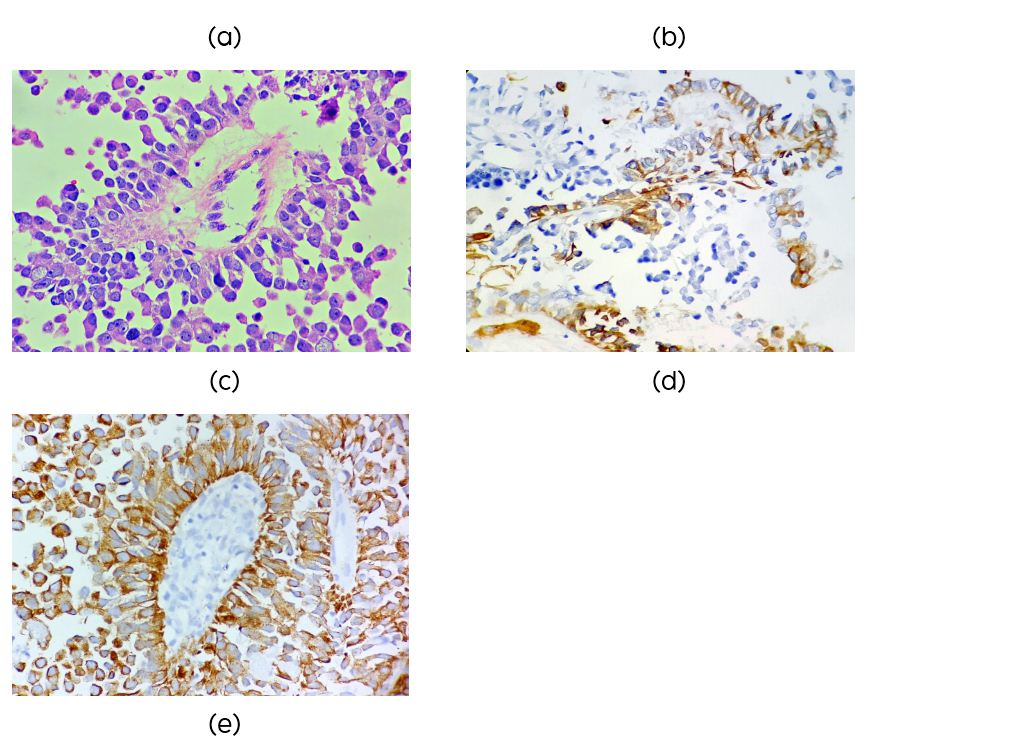
Figures 1: (a) Enhancing solid and cystic mass in IIIrd ventricle and penial gland (MRI); (b, c) Low and high power view showing papillary architecture (H&E ×10, ×40); (d) Focal positivity for GFAP (×40); (e) Diffuse positivity for cytokeratin (×40).
Case 2
A 22-years-old female with a complaints of diplopia since two months, had a midbrain lesion which had displaced the tectal plate on MRI (Figure 4). On squash the tumor showed clusters with papillary configuration (Figure 2a), suggestive of a tumor of glial origin favoring ependymoma.
On microscopy the cellular tumor displayed papillary architecture, perivascular pseudo rosettes and sheets of tumors cells infiltrating fibrillary matrix. Cells around the vessels were cuboidal to columnar and showed stratification, had acidophilic cytoplasm, round to oval nuclei with evenly dispersed chromatin (Figure 2b). There was no necrosis endothelial proliferation or significant mitotic activity or atypia. Possibilities considered were papillary ependymoma, choroid plexus carcinoma, papillary tumor of pineal region and metastatic carcinoma. On immunohistochemistry the tumor cells were diffusely positive for pancytokeratin (Figure 2c), S100 (Figure 2d), Neuron specific enolase (NSE) and Vimentin (Figure 3a, b), focally positive for GFAP, and EMA negative. Mib-1 index was 1%. IHC was compatible with the diagnosis of PTPR. The tumor recurred six months post-surgery and radiotherapy.
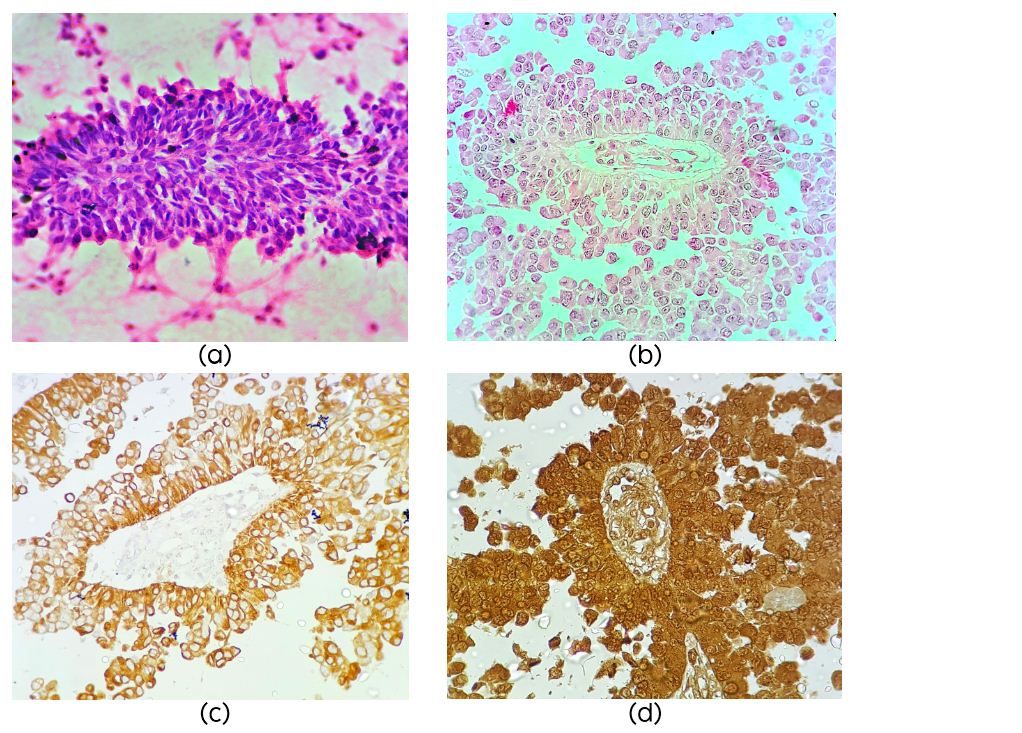
Figures 2: (a) Squash smear showing papillae (H&E ×40); (b) Papillary architecture (H&E ×40); (c) Diffuse positivity for cytokeratin (×40); (d) Strong S100 positivity (×40).
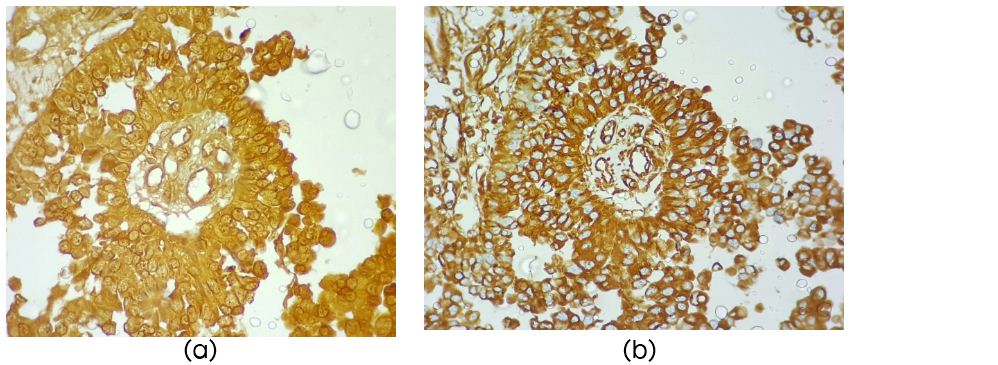
Figure 3: (a) Diffuse strong NSE positivity (×40); (b) Diffuse positivity for vimentin (×40).
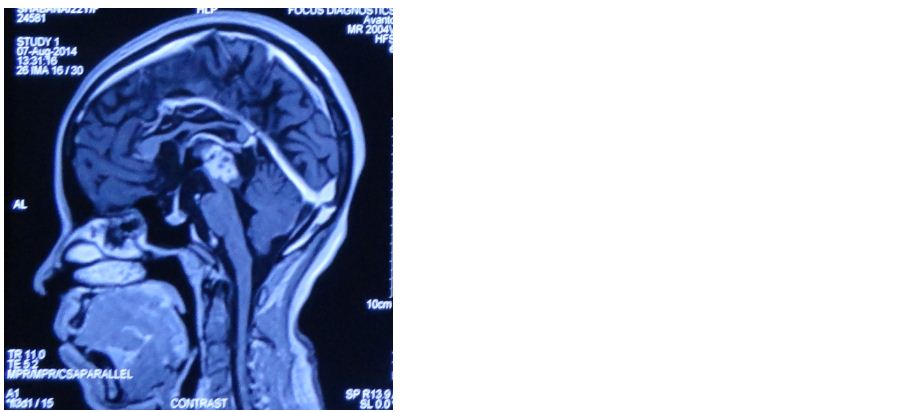
Figure 4: Faintly hyperintense tumor displacing the tectal plate (MRI).
Discussion
The PTPR affects mostly young adults in their thirties and also children [3, 4]. There is no sex prevalence. Jouvet et al. first described six PTPR patients in 2003, since then PTPR has been increasingly recognised and some past diagnoses have also been revised [5]. Its biological behavior corresponds to a WHO grade II or III neoplasm but exact histological grading criteria are yet to be defined.
The common symptom at presentation is increase of intracranial pressure, Other symptoms include Parinaud’s syndrome, ataxia and isolated diplopia as seen in these two patients.
PTPR at presentation are usually 2 - 4 cm [2], well-circumscribed and sometimes cystic [6, 7]. PTPRs have two morphological variants: papillary and solid papillary growth patterns [3]. True rosettes and perivascular pseudorosettesare also observed. The tumor cells usually have a columnar to cuboidal shape, with a defined cytoplasmic membrane and epithelioid nucleus. The cells which are vacuolated are sometimes show partial positivity for periodic acid-schiff reaction [4, 8]. There may be necrosis and moderate mitotic activity. PTPR shows strong reactivity for cytokeratin 18 [9, 2]. There is weak or absent expression of CK7, CK20, CK5/6.Epithelial membrane antigen (EMA) expression maybe absent as in the above cases, weak, dot-like or ring-like. E-cadherin is not expressed whereas claudins 1 and 3 are expressed focally [3]. S100 protein (S100P) and vimentin are positive. Immunoreactivity for glial fibrillary acidic protein is rare. Neuroendocrine markers (neuron specific enolase, chromograninA, synaptophysin) maybe present. Neurofilament protein expression have been described in few reports [10, 11], whereas microtubule-associated protein 2 staining is found in the majority of cases.
The differential diagnosis includes most commonly papillary ependymoma, choroid plexus papilloma, papillary meningioma, and metastatic papillary carcinoma. Based on their distinct morphological features and immunophenotypes these entities can be excluded. Rare cases of pineal parenchymal tumors with papillary features have been reported, including a papillary pineocytoma [12] and amalignant pineocytoma with prominent papillary features [13]. In contrast to pineal parenchymal tumors, PTPRs usually lack expression of retinal S-antigen, as well as neurofilament protein, and show prominent epithelial features, including cytokeratin, S100 and vimentin immunoreactivity [14].
It is hypothesized that these lesions originate from the specialisedcytokeratin-positive and nestin-positive ependymal cells of the subcommissural organ (SCO) based on ultrastructural and immunohistochemical features [1, 3]. Supporting this hypothesis, a microarray study has shown high expression of genes also expressed in the rodent Sub commissural organ [12].
Comparative genomic hybridization analysis of five cases of PTPR showed frequent losses on chromosomes 10 and 22, and gainson chromosomes 4, 8, 9 and 12 [15]. A microarray study shows high expression of SPDEF, KRT18, and genes encoding proteins reportedto be expressed in the SCO, namely ZFH4, RFX3, TTR and CGRP [6].
PTPRs are known to recur locally, however rare cases of spinal metastasis and meningeal spread have also been reported [2, 4]. PTPRs at younger age i.e. < 30 years of age were reported to have high Ki-67 but were not aggressive and did not negatively impact on progression-free and overall survival [2]. However, in case 2 who was 22-years-old had Mib-1 index of 1%.
Conclusion
PTPR is a rare tumor, which has high rate of recurrence and potential for CSF dissemination or meningeal spread. It should be included in the differential diagnosis in cases where the tumor is arising in the vicinity of pineal region showing papillary architecture.
Acknowledgement
The Departments of Pathology, Radiology, and Neurosurgery, KIMS hospital, Secunderabad, India.
Conflicts of interest
The authors declare no conflicts of interest.
References
[1] FèvreMontange M, Champier J, Szathmari A, Wierinckx A, Mottolese C, et al. Microarray analysis reveals differential gene expression patterns in tumors of the pineal region. J Neuropathol Exp Neurol. 2006; 65(7):675–684.
[2] FèvreMontange M, Hasselblatt M, Figarella-Branger D, Chauveinc L, ChampierJ, et al. Prognosis and histopathologic features in papillary tumors of the pineal region: A retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol. 2006; 65(10):1004–1011.
[3] FèvreMontange M, Vasiljevic A, Bergemer Fouquet AM, Bernier M, Champier J, et al. Histopathologic and ultrastructural features and claudin expression in papillary tumors of the pineal region: A multicenter analysis. Am J Surg Pathol. 2012; 36(6):916–928.
[4] Marcol W, Kotulska K, Grajkowska W, Gołka D, Właszczuk P, et al. Papillary pineocytoma in child: A case report. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2007; 151(1):121–123.
[5] Jouvet A, Fauchon F, Liberski P, Saint-Pierre G, Didier-Bazes M, et al. Papillary tumor of the pineal region. Am J Surg Pathol. 2003; 27(4):505–512.
[6] Kern M, Robbins P, Lee G, Watson P. Papillary tumor of the pineal region–a new pathological entity. Clin Neuropathol. 2006; 25(4):185–192.
[7] Patel SK, Tomei KL, Christiano LD, Baisre A, Liu JK. Complete regression of papil-lary tumor of the pineal region after radiation therapy: Case report and review of the literature. J Neurooncol. 2012; 107(2):427–434.
[8] Kuchelmeister K, Hugens-Penzel M, Jodicke A, Schachenmayr W. Papillary tumour of the pineal region: Histodiagnostic considerations. Neuropathol Appl Neurobiol. 2006; 32(2):203–206.
[9] Rickard KA, Parker JR, Vitaz TW, Plaga AR, Wagner S, et al. Papillary tumor of the pineal region: two case studies and a review of the literature. Ann Clin Lab Sci. 2011; 41(2):174–181.
[10] Cykowski MD, Wartchow EP, Mierau GW, Stolzenberg ED, Gumerlock MK, et al. Papillary tumor of the pineal region: Ultrastructural study of a case. Ultra-struct Pathol. 2012; 36(1):68–77.
[11] Nakamura H, Makino K, Kochi M, Nakazato Y, Kuratsu J. Successful treatment of neoadjuvant therapy for papillary tumor of the pineal region. Brain Tumor Pathol. 2009; 26(2):73–77.
[12] Vaquero J, Coca S, Martinez R, Escandon J. Papillary pineocytoma: Case report. J Neurosurg. 1990; 73(1):135–137.
[13] Trojanowski JQ, Tascos NA, Rorke LB. Malignant pineocytoma with prominent papillary features. Cancer. 1982; 50(9):1789–1793.
[14] Fèvre Montange M, Vasiljevic A, Champier J, Jouvet A. Papillary tumor of the pineal region: Histopathological characterization and review of the literature. Neurochirurgie. 2015; 61(2-3):138–142.
[15] Hasselblatt M, Blümcke I, Jeibmann A, Rickert CH, Jouvet A, et al. Immunohistochemical profile and chromosomal imbalances in pap-illarytumours of the pineal region. Neuropathol Appl Neurobiol. 2006; 32(3):278–283.