Full Text
Adult-Onset Still’s Disease (AOSD) was first described by Bywaters in 1971. Still disease is a systemic inflammatory disorder of unknown etiology. AOSD is uncommon, with an estimated prevalence lower than 1 case per 100,000 people. Patients usually present with a high spiking fever, arthralgia or arthritis, sore throat, transient distinctive salmon-colored bumpy rash, lymphadenopathy, Hepatosplenomegaly and serositis [1]. Patients may or may not have all of the above symptoms at initial presentation. The disease is considered as a diagnosis of exclusion. Levels of the iron-binding protein ferritin may be elevated with this disorder. AOSD may present in a similar manner to other inflammatory diseases and to autoimmune diseases, which must be ruled out before making the diagnosis. Prognosis is usually favorable but manifestations of the disease affecting the lungs, heart, or kidney may occasionally cause severe life-threatening complications [2].
Case report
A 24-year-old male presented to the emergency department with complaints of having had sore throat, joint pain, and a spiking fever for 5 days. The rest of the review of the systems was negative. He had no significant medical history, was not taking any medication, and had no significant family history. Vital signs at admission were a temperature of 38.8°C, heart rate of 92 beats per minute, respiratory rate of 20 breaths per minute, and blood pressure of 126/74 mm Hg. Physical examination was normal except for maculopapular skin rash, dermatological opinion was taken. They were in favour of seborrheic dermatitis. Cardiovascular, respiratory system, abdominal examination and the genitourinary examination was normal. Total white blood cell count, bands, C- reactive protein level, erythrocyte sedimentation rate (ESR), and liver function test were elevated. The chest radiograph was normal. Urinalysis, urine culture, and 3 sets of blood cultures were negative and he underwent further work-up. An infectious disease workup was done, and was negative. Bone marrow biopsy was done which was normal.
At this stage an autoimmune disorder was considered. Antinuclear antibodies (ANA) test was done which was negative. Anti-neutrophil cytoplasmic antibodies (ANCA) ANCA, perinuclear ANCA (p-ANCA) was done which was negative to rule out vasculitis. RA factor was negative, serum ferritin was >3000. The patient was evaluated by a rheumatologist and was noted to fulfil the proposed diagnostic criteria of AOSD. He was started on appropriate therapy.
Discussion
AOSD is a rare inflammatory disorder that usually affects young adults, although it can also be seen among the geriatric population. There is a slight preponderance among women. The mechanisms underlying AOSD are not completely understood to date, and the possibility of a genetic association remains inconclusive. Although AOSD pathogenesis is not known, it is considered as a member of the expanding group of the autoinflammatory disorders [2]. These diseases are caused by aberrancies in the innate inflammatory pathways, and increased release of active IL-1β is considered as a major event in their pathogenesis. Of late, in a small series of patients with AOSD, increased serum levels of proinflammatory cytokines (IL-6, IFNγ, IL-18, and IL-1β) have been reported [3]. NLRP3 inflammasome has been identified as the key intracellular platform for the maturation of IL-1β, and dysregulation of its function has been shown to have major role in the pathophysiology of autoinflammatory, metabolic, and certain autoimmune diseases [6]. There are several infectious agents related to AOSD onset reported in the literature and based on concurrent elevation of serology markers. These include Epstein–Barr; parvovirus B19; cytomegalovirus; human herpes virus 6, human immunodeficiency virus; coxsackievirus; mumps; rubella, echovirus, Hepatitis A, B, and C viruses; campylobacter jejuni; chlamydia pneumonia, and Mycoplasma pneumonia. Distinguishing between infections related AOSD with chronic self-perpetuating inflammation versus long-lasting infection with chronic persistence of the infectious agent can be difficult.
Clinically, most of the patients with AOSD present with fever, sore throat, arthralgia, arthritis, and/or skin rash, but some patients may also have present with conditions such as lymphadenopathy, hepatosplenomegaly, and/ or serositis. The fever, typically higher than 39°C, starts suddenly and could present as UO alone. Joint pains occur in two thirds of patients. The arthralgia starts concomitantly with fever, involves any joint, and may migrate at the beginning and become more stable during the course of the disease. The skin rash consists of small discrete, nonpruiritic, salmon-pink macules or maculopapules, which are transient and mainly visible during fever.
AOSD is a diagnosis of exclusion. Differential diagnoses include infections, such as endocarditis and deep-seated occult infections, or neoplastic etiology, especially lymphomas and autoimmune diseases like vasculitis and polymyositis. These conditions should be excluded before AOSD is diagnosed. Eight different [4-10] sets of diagnostic criteria have been proposed for clinical diagnosis of AOSD3, The most widely used and validated criteria are those of Yamaguch (Table 1). The Yamaguchi criteria [4] have a sensitivity of 96.2% and specificity of 92.1%. A study by Crispin et al. showed that the Yamaguchi criteria had a positive predictive value of 70.3% and negative predictive value of 95.2% among their study population.
Table 1: Yamaguchi criteria.
|
Major criteria
|
Minor criteria
|
|
Fever of at least 39oC for at least one week
|
Sore throat
|
|
Arthralgias or arthritis for at least two weeks
|
Lymphadenopathy
|
|
Nonpruritic salmon colored rash (usually over trunk or extremities while febrile)
|
Hepatomegaly or splenomegaly
|
|
Leukocytosis ( 10,000/microL or greater), with granulocyte predominance
|
Abnormal liver function tests
|
| |
Negative tests for antinuclear antibody and rheumatoid factor
|
The laboratory investigations can show an increased systemic inflammatory response, such as high white blood cell count (usually 10,000 to 15,000 with more than 80% granulocytes), thrombocytosis; anemia; ESR; elevated C-reactive protein, and significant increase in serum ferritin level. ESR level could be >100 mm in some cases. Coagulation abnormalities, abnormal liver function tests, and raised lactic dehydrogenase level are also seen. A high level of ferritin seems to be characteristic of AOSD and is seen in nearly 70% of cases. Ferritin levels in AOSD are usually higher than those found in other autoimmune or inflammatory disorders. Serum ferritin levels of >3000 ng/mL have been most commonly observed among patients with AOSD; glycosylated ferritin level drops to ≤20% in patients with AOSD.
Natural history of AOSD has been described commonly in 3 different patterns. Monocyclic, or self-limited, systemic pattern is usually associated with a single flare and complete remission is achieved within 2 to 4 weeks in 19% to 44% of cases. Intermittent, or polycyclic, pattern is seen in 10% to 41% of cases and it usually presents with recurrence of systemic or articular flares separated by periods of remission lasting from 2 weeks to 2 years. The chronic evolution form more frequently is articular than systemic and is seen in 35% to 67% of cases.
The treatment of AOSD is mainly centered on the use of nonsteroidal anti-inflammatory drugs, steroids, and disease-modifying antirheumatic drugs. No randomized controlled trial has been performed to evaluate treatments for AOSD; nonsteroidal anti-inflammatory drugs and prednisone are used as firstline agents in the treatment of AOSD. After the diagnosis is established, corticosteroids are usually required for symptom control and response is often dramatic. Patients with visceral involvement may achieve a response with an intravenous infusion of high-dose methylprednisolone [11]. Methotrexate is effective in controlling disease activity and allows for steroid dose sparing in AOSD. Other commonly used medications include hydroxychloroquine, penicillamine, azathioprine, methotrexate, etanercept, anakinra, cyclophosphamide, adalimumab, rituximab, and infliximab.
Newer drugs target interleukin-1 (IL-1), particularly IL-1β. A randomized, multicenter trial reported better outcomes in a group of 12 patients treated with anakinra than in a group of 10 patients taking other disease-modifying antirheumatic drugs [Figure 1]. Other anti-IL1β drugs are being developed, including canakinumab and rilonacept.
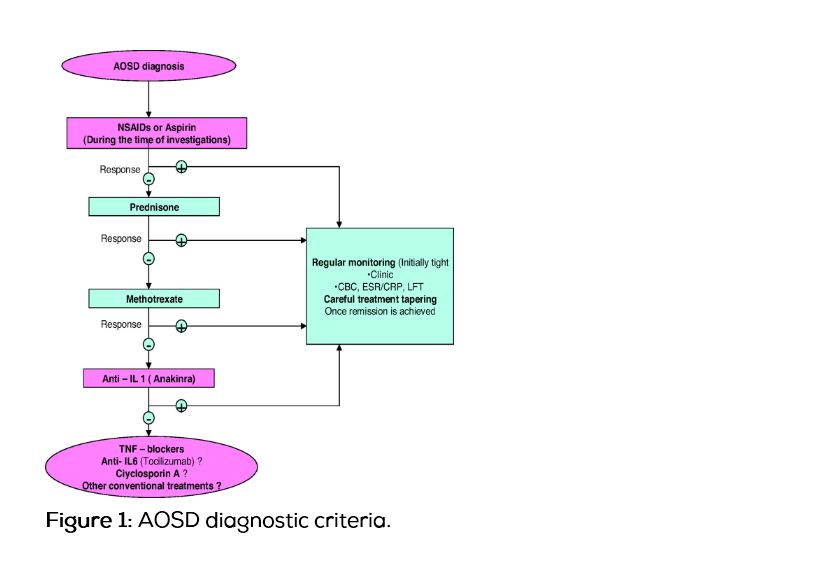
Conclusion
AOSD is a rare autoimmune disorder. The most characteristic clinical presentation of this condition is fever, arthralgia or arthritis, sore throat, and skin rash. Fever of unknown origin is one of the most common presentations of AOSD. Diagnosis is based on exclusion of inflammatory and neoplastic diseases and no definite serologic marker is available at present. It is still controversial as to whether the infections found in conjunction with AOSD are merely “contemporaneous” or if they may indeed be triggering factors of AOSD.
Conflict of interest
The authors declare no conflict of interest.
References
1. Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease.Ann Rheum Dis. 2006; 65(5):564–572.
2. Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010; 140(6):784–790.
3. Kötter I, Wacker A, Koch S, Henes J, Richter C, et al. Anakinra in patients with treatment-resistant adult-onset Still's disease: four case reports with serial cytokine measurements and a review of the literature. Semin Arthritis Rheum. 2007; 37(3):189–197.
4. Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992; 19(3):424–430.
5. Cush JJ, Medsger TA, Christy WC, Herbert DC, Cooperstein LA. Adult onset Still's disease: clinical course and outcome. Arthritis Rheum. 1987; 30(2):186–194.
6. Fautrel B, Zing E, Golmard JL, Le Moel G, Bissery A, et al. Proposal for a new set of classification criteria for adult-onset Still disease. Medicine (Baltimore). 2002; 81(3):194–200.
7. Khan MF, Delaire M. Maladies de Still de l adulte. In: Khan MF, Peltier AP, Meyer O, Piette JC, eds. Les Maladies Systemiques. Paris: Flammarion; 1991: 231.
8. Calabro JJ, Londino AV. Adult onset Still's disease. J Rheumatol. 1986; 13(4):827–828.
9. Goldman JA, Beard MR, Casey HL. Acute febrile juvenile rheumatoid arthritis in adults: cause of polyarthritis and fever. South Med J. 1980; 73(5):555–563.
10. Reginato AJ, Schumacher HR, Baker DG, O'Connor CR, Ferreiros J. Adult onset Still's disease: experience in 23 cases and literature review with emphasis on organ failure. Semin Arthritis Rheum. 1987; 17(1):39–57.
11. Bisagni-Faure A, Job-Dedlandre C, Menkes CJ. Intravenous methylprednisolone pulse therapy in Still's disease. J Rhumatol 1992; 19(9):1487–1488.