Full Text
Introduction
Pancreatic neuroendocrine tumors (pancreatic NETs) are rare neoplasms with an estimated incidence of ≤1 per 100,000 population per year, accounting for less than 3% of all primary pancreatic tumors [1]. These tumors may occur sporadically or in association with hereditary syndromes such as multiple endocrine neoplasia type 1 (MEN-1), neurofibromatosis type 1 (NF-1), tuberous sclerosis, and Von Hippel–Lindau syndrome [2]. Pancreatic NETs are broadly classified into functioning (F-NET), which secrete bioactive peptides leading to characteristic clinical syndromes, and non-functioning NETs (NF-NET), which may secrete peptides such as pancreatic polypeptide but do not produce overt clinical symptoms [3].
Given their heterogeneity in clinical presentation, biological behavior, and therapeutic response, early diagnosis and individualized management plays a crucial role in improving outcomes. Advancements in cross-sectional imaging, endoscopic ultrasound, functional imaging, and histopathological grading have significantly improved diagnostic precision and treatment planning in recent years [4–7].
In this case series, we retrospectively analyzed eight patients with pancreatic NETs managed in the Department of Endocrinology. Four patients had functioning tumors (insulinomas), all presenting with symptomatic hypoglycaemia, confirmed by low plasma glucose with inappropriately elevated insulin and C-peptide levels. The objective of this report is to describe their clinical profiles, diagnostic approaches, management strategies, and outcomes to highlight the heterogeneity of pancreatic NETs and the importance of comprehensive multidisciplinary care.
Case presentations
We retrospectively collected data from eight patients diagnosed with pancreatic neuroendocrine tumors (NETs) in the Department of Endocrinology, Krishna Institute of Medical Sciences Hospital, between March 2022 and February 2024. Clinical presentations, biochemical profiles, imaging findings, histopathology, staging, treatments, and follow-up outcomes were analyzed. Data were obtained from inpatient medical records, outpatient follow-up documentation, and laboratory databases.
Patient demographics such as age, sex, and presenting symptoms were reviewed along with biochemical evaluation, including plasma glucose levels assessed in correlation with simultaneously measured insulin and C-peptide concentrations to confirm endogenous hyperinsulinism in functioning NETs (insulinomas). Imaging modalities included contrast-enhanced computed tomography (CECT), Ga-68 DOTATATE PET/CT, Ga-68 Exendin PET/CT where indicated, and endoscopic ultrasound (EUS) for localization and staging. Histopathological analysis provided tumor grade, Ki-67 (MIB-1) proliferation index, and immunohistochemical characteristics. Therapeutic interventions included surgical resection, somatostatin analogues (octreotide LAR), and peptide receptor radionuclide therapy (PRRT) for advanced or unresectable disease. Clinical outcomes were monitored in relation to symptom resolution, disease progression, and long-term follow-up.
A total of eight pancreatic NET cases were evaluated, comprising four functioning and four non-functioning tumors. The study was approved by the Institutional Ethics Committee, and informed consent was obtained from all participants.
Case 1
A 30-year-old female with BMI 22.7 kg/m² presented with episodes of sweating and palpitations. Random blood glucose was 43 mg/dl, with markedly elevated insulin (109 µU/ml) and C-peptide (7.2 ng/ml), confirming endogenous hyperinsulinism. CECT abdomen was normal, but Ga-68 Exendin PET-CT localized a 3 × 4 cm hypermetabolic lesion (SUVmax 8.3) in the pancreatic tail. She underwent distal pancreatectomy, and histopathology confirmed WHO grade 1 NET with MIB-1 index of <3%, T2N0M0 (stage 2, AJCC 9). Postoperatively, the patient had no further hypoglycaemic episodes.
Case 2
A 36-year-old female with a BMI of 26 kg/m² presented with recurrent convulsions attributable to hypoglycaemia, with lowest documented RBS at 30 mg/dl. Insulin (39.9 µU/ml) and C-peptide (6.67 ng/ml) levels confirmed endogenous hyperinsulinism. CECT showed an exophytic lesion in the pancreatic tail, further characterized by EUS as a 1.2 × 1.3 cm hypoechoic lesion. The patient underwent laparoscopic distal pancreatectomy. Histopathology confirmed a WHO grade 1 NET with MIB-1 index of 2%, T1N0M0 (stage 1, AJCC 9). On follow-up, no further hypoglycaemic episodes were reported.
Case 3
A 45-year-old female with a BMI of 34.4 kg/m² presented with recurrent episodes of sweating and altered sensorium, with random blood glucose documented at 29 mg/dl. On examination there was grade 4 Acanthosis nigricans with skin tags as shown in figure 1. Laboratory investigations revealed elevated insulin (31.8 µU/ml) and C-peptide (2.28 ng/ml), confirming endogenous hyperinsulinism and fulfilling Whipple’s triad. Contrast-enhanced CT and Ga-68 DOTATATE PET/CT were normal, but endoscopic ultrasound detected a 1.8 × 1.6 cm hypoechoic lesion in the pancreatic body as shown in figure 2. Histopathology confirmed a WHO grade 1 neuroendocrine tumor with an MIB-1 index of 2%, T1N0M0 (stage 1, AJCC 9). The patient underwent endoscopic ultrasound-guided radiofrequency ablation (EUS-RA) at 30W for 12 seconds per spot. Post procedure there was complete resolution of hypoglycaemic episodes. No recurrence of symptoms on follow-up.

Figure 1: Case 3 with grade 4 acanthosis in insulinoma.
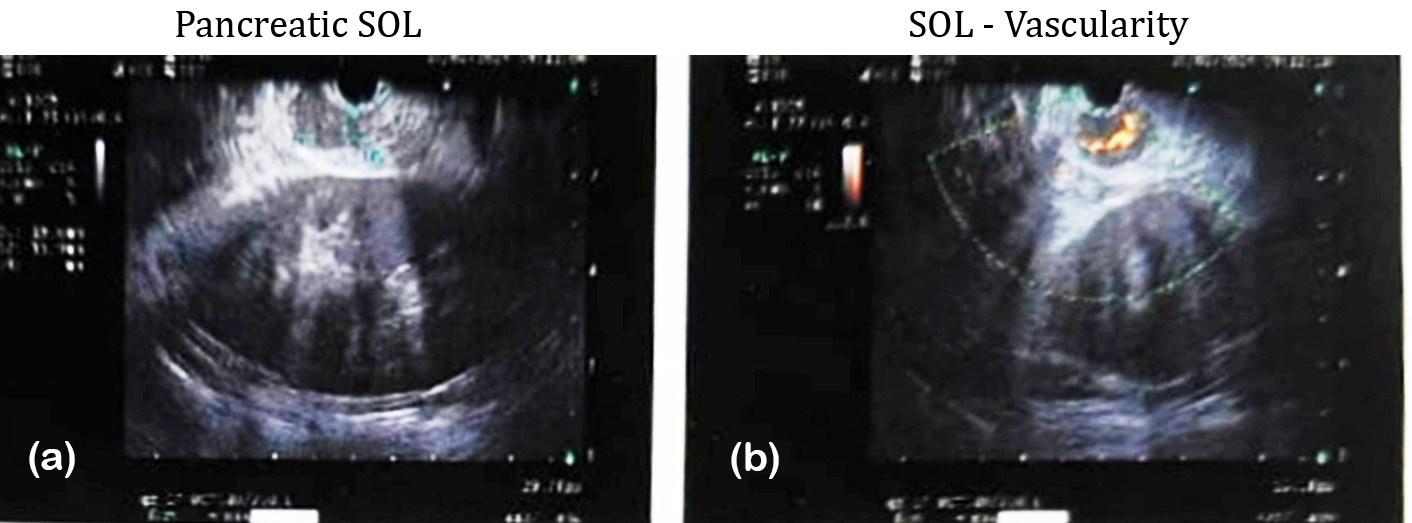
Figure 2: Case 3, endoscopic ultrasound.
Case 4
A 46-year-old male (BMI 28 kg/m²) presented with recurrent episodes of sweating and fatigue, with documented blood glucose of 34 mg/dl. Laboratory evaluation showed elevated insulin (60.7 µU/ml) and C-peptide (5.69 ng/ml), confirming endogenous hyperinsulinism. CECT abdomen revealed a 1.9 × 1.6 cm lesion in the uncinate process of the pancreas, with no evidence of regional or distant disease on nuclear imaging. The patient underwent minimally invasive enucleation of the insulinoma. Histopathology confirmed a WHO grade 1 NET with an MIB-1 index <3%, staged as T1N0M0 (AJCC 9). On follow-up, he remained asymptomatic with complete resolution of hypoglycaemic episodes.
The presenting features and outcome data of the 4 functioning neuroendocrine tumor cases are enumerated in table 1 and 2 respectively.
Table 1: Clinical and laboratory features of cases with functioning neuroendocrine tumor.
|
Age
(yrs)/ Gender
|
Presenting
complaints
|
BMI
(kg/m2)
|
RBS
(mg/dl)
|
Insulin
(μU/ml)
(NR=2.6-24.9)
|
c-peptide
(ng/ml)
(NR=0.81-3.85)
|
Diagnosis
|
|
30/F
|
Sweating,
palpitations
|
22.7
|
43
|
109
|
7.2
|
Endogenous hyperinsulinism
|
|
36/F
|
Convulsions
|
26
|
30
|
39.9
|
6.67
|
Endogenous hyperinsulinism
|
|
45/F
|
Episodes of sweating, altered sensorium
|
34.4
|
29
|
31.8
|
2.28
|
Endogenous
hyperinsulinism
|
|
46/M
|
Sweating,
Fatigue
|
28
|
34
|
60.7
|
5.69
|
Endogenous hyperinsulinism
|
Table 2: Functioning NETs cases localization methods, treatment and follow-up.
|
CECT abdomen
|
Nuclear
scan
|
Endoscopic ultrasound
|
HPE
|
MIB-1
|
Stage
AJCC9
|
First line treatment
|
Follow-up
|
|
Normal
|
68Ga-Exendin
PET-CT:
3x4 cms lesion in tail (8.3-
SUVmax)
|
-
|
NET
WHO-
Grade1
|
<3%
|
T2N0M0
(stage2)
|
Distal pancreatectomy
|
No further hypoglycaemic episodes
|
|
Normal
|
Exophytic
Lesion in tail of pancreas
|
1.2x1.3 cms hypoechoic lesion in tail of pancreas
|
NET
WHO-
Grade1
|
2%
|
T1N0M0
(stage 1)
|
Laparoscopic
Distal pancreatectomy
|
No further hypoglycaemic episodes
|
|
Normal
|
68Ga-
DOTATATE
PET-CT:
Normal
|
1.8x1.6cms
Lesion in body of pancreas
|
NET
WHO-Grade1
|
2%
|
T1N0M0
(Stage1)
|
EUS guided Radio-
Frequency
ablation
|
No further hypoglycaemic episodes
|
|
1.9x1.6cms
Lesion in uncinate process of pancreas
|
-
|
-
|
NET
WHO-Grade1
|
<3%
|
T1N0M0
(stage1)
|
Minimally invasive enucleation of insulinoma
|
No further hypoglycaemic episodes
|
Non-functioning pancreatic neuroendocrine tumors
Case 5
A 37-year-old female (BMI 24.5 kg/m²), known to have Multiple Endocrine Neoplasia type 1 (MEN-1) with a history of pituitary macroadenoma and acromegaly, presented with backache. CT abdomen revealed an ill-defined isodense-to-hypodense lesion involving the head and uncinate process of the pancreas, along with multiple enlarged lymph nodes encasing the extrahepatic portal vein. Ga-68 DOTATATE PET/CT demonstrated multiple pancreatic lesions with metastatic deposits in the liver and regional lymph nodes. Histopathology confirmed a WHO grade 2 neuroendocrine tumor with a mitotic rate of 4/mm², staged as T3N1M1 (stage IV; AJCC 9). The patient was kept under active surveillance and continued to have stable disease on imaging at 2-year follow-up.
Case 6
A 53-year-old male (BMI 20 kg/m²) presented with abdominal pain. CT imaging revealed a 2 × 3.4 cm lesion in the uncinate process of the pancreas, along with a metastatic lesion in the liver. Ga-68 DOTATATE PET/CT confirmed pancreatic and hepatic involvement with nodal metastases (figure 3). Histopathology showed a WHO grade 2 NET with a mitotic rate of 8 per 10 HPF, staged as T2N1M1 (stage IV;AJCC-9). The patient underwent surgical resection of the liver lesion and was initiated on octreotide LAR therapy. Due to progressive disease on follow-up imaging, he subsequently received three cycles of 177Lu-DOTATATE PRRT (150 mCi). Post-treatment Ga-68 DOTATATE PET/CT demonstrated a reduction in lesion size and metabolic activity as shown in figure 4, and he achieved 2 years of progression-free survival following therapy.
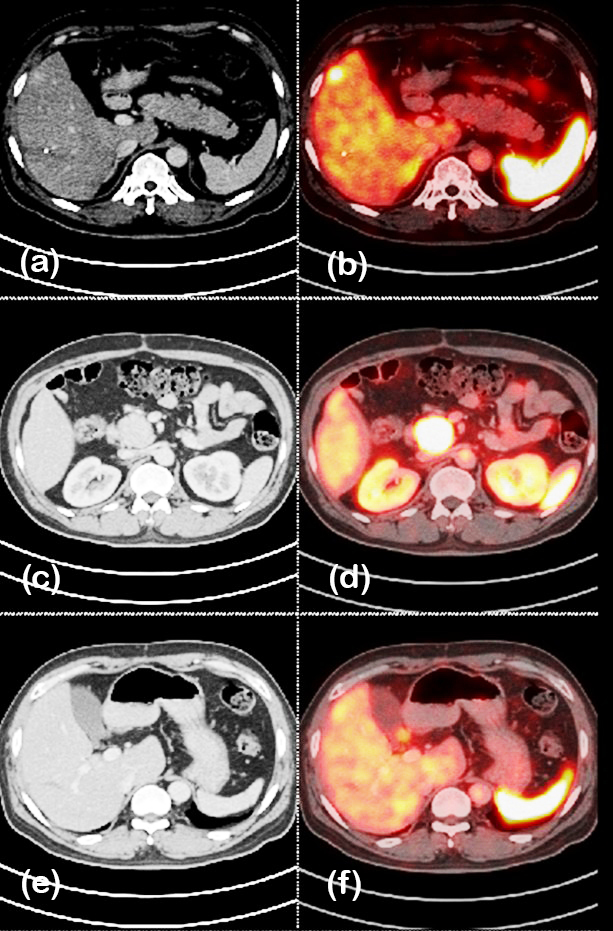
Figure 3: Case 6- 68Ga DOTATATE PET/CT before PRRT showing lesion in pancreas.
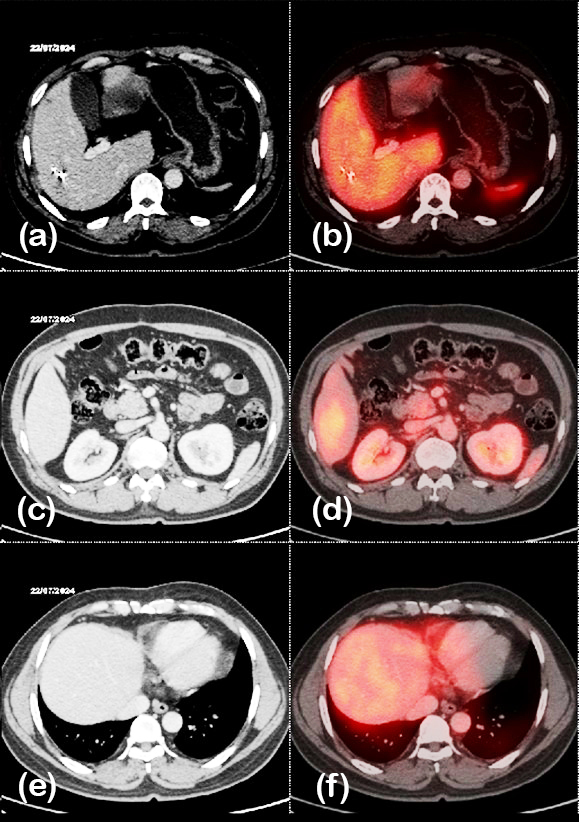
Figure 4: Case 6- 68Ga DOTATATE PET/CT after PRRT, showing decrease in size and uptake of pancreatic lesion.
Case 7
A 56-year-old male (BMI 19 kg/m²) with clinical features suggestive of Neurofibromatosis type 1 (NF-1) including multiple cutaneous neurofibromas, café-au-lait macules, and palmar freckling (Figure 5) presented with jaundice. Ultrasonography of the abdomen revealed a 1.2 × 1.5 cm mass in the periampullary region of the pancreas. Ga-68 DOTATATE PET/CT localized a corresponding somatostatin receptor–avid 1.9 × 1.5 cm lesion without evidence of metastasis (figure 6). Histopathological examination confirmed a WHO grade 1 neuroendocrine tumor with a mitotic rate <2/mm², staged as T1N0M0 (stage I; AJCC-9). The patient underwent surgical resection of the periampullary mass and remained disease-free at 2-year follow-up.

Figure 5: Case 6, NF-1 with neurofibromas and café-au-lait macules.
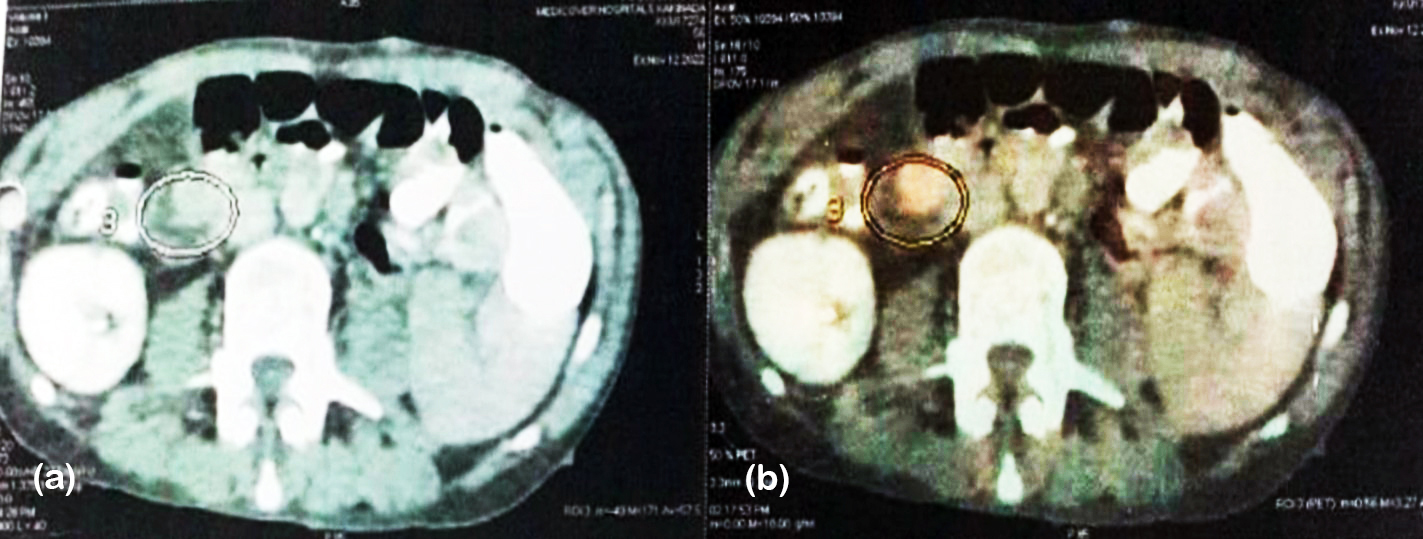
Figure 6: PET CT scan of NF-1 case showing metabolically active lesion in peri-ampullary region measuring approximately 1.9x1.5x1.5 cms.
Case 8
A 63-year-old male (BMI 18 kg/m²) presented with progressive jaundice. CT abdomen revealed a 4.3 × 3 cm mass involving the head and uncinate process of the pancreas. Ga-68 DOTATATE PET/CT demonstrated somatostatin receptor–avid lesion in the pancreatic head and duodenum, with involvement of regional lymph nodes. Histopathology confirmed a WHO Grade 2 neuroendocrine tumor with a mitotic rate of 5 per 10 HPF, staged as T3N1M0 (stage III;AJCC-9). The patient underwent pancreaticoduodenectomy and was subsequently initiated on octreotide LAR therapy every 4 weeks. Due to disease progression, he later received four cycles of 177Lu-DOTATATE PRRT (150 mCi). Follow-up Ga-68 DOTATATE PET/CT demonstrated reduced lesion size and markedly decreased tracer uptake, and he achieved 2 years of progression-free survival after therapy. The presenting features and outcome data of the 4 non-functioning neuroendocrine tumor cases are enumerated in table 3 and 4 respectively.
Table 3: Clinical features, imaging of cases non-functioning pancreatic NETs.
|
Age
(yrs)/ Gender
|
Presenting
complaints
|
BMI
(kg/m2)
|
Associated
syndrome
|
Imaging findings
|
|
37/F
|
backache
|
24.5
|
MEN-1
(h/o pituitary macroadenoma-Acromegaly)
|
CT abdomen: ill-defined iso-hypodense lesion in head and uncinate process of pancreas, multiple enlarged lymph nodes encasing extrahepatic portal vein
|
|
53/M
|
Pain abdomen
|
20
|
-
|
CT abdomen- 2x3.4 cms lesion in uncinate process of pancreas and liver lesion
|
|
56/M
|
Jaundice
|
19
|
NF-1 (cutaneous neurofibromas,
café-au-lait macules, palmar freckling -present)
|
USG abdomen- 1.2x1.5 cms mass in peri-ampullary region of pancreas
|
|
63/M
|
Jaundice
|
18
|
-
|
CT abdomen: 4.3x3 cms mass in head and uncinate process of pancreas
|
Table 4: Nuclear scan, histopathology, staging and treatment in NF-NETs.
|
Ga-68 DOTATATE
PET/CT
|
HPE
|
Mitotic
Rate
|
Staging
AJCC9
|
First line
treatment
|
Oct
LAR
|
At 12 weeks
|
PRRT
|
Follow-up
|
|
Multiple lesions in pancreas, with metastasis in liver and LNs
|
NET
WHO-
Grade2
|
4/mm2
|
T3N1M1
(Stage4)
|
Surveillance
|
-
|
Stable disease
|
-
|
Stable disease at 2 years
|
|
Lesion in the pancreas, liver
and LNs
|
NET
WHO-grade2
|
8/10 hpf
|
T2N1M1
(stage4)
|
Resection of lesion in the liver
|
Every 4 weekly
|
Progressive
|
3 cycles
|
Decrease in size and uptake of lesion in Ga-68 DOTATATE PET/CT and 2 years
PFS
|
|
1.9x1.5cms periampullary mass
|
NET
WHO-grade1
|
<2/mm2
|
T1N0M0
(stage1)
|
Surgical resection of periampullary mass
|
-
|
-
|
-
|
2 years
DFS
|
|
Lesion in head of pancreas & duodenum, LNs
|
NET
WHO-garde2
|
5/10 hpf
|
T3N1M0
(stage3)
|
Pancreatico-
duodenectomy
|
Every 4
weekly
|
Progressive
|
4 cycles
done
|
Decrease in size and uptake of lesion in Ga-68 DOTATATE
PET/CT and 2 years
PFS
|
Discussion
Optimal management of pancreatic NETs depends on tumor functionality, grade, and stage. Functioning tumors such as insulinomas cause hypoglycaemia due to autonomous insulin secretion. When clinical suspicion for insulinoma is high, localization strategies include functional imaging such as glucagon-like peptide-1 receptor PET/CT, endoscopic ultrasound, and, when required, selective arterial calcium stimulation with hepatic venous sampling (ASVS), which remains the diagnostic gold standard [13]. Medical management with diazoxide or somatostatin analogues may be necessary in patients who are not candidates for surgery. Prognosis is generally favorable after surgical resection; however, ongoing surveillance is essential, particularly in cases of malignant or metastatic disease [14]. Surgical excision or minimally invasive ablation aims to achieve cytoreduction, relieve symptoms, and prevent recurrence. Insulinomas typically demonstrate benign histological features and excellent long-term survival outcomes [2, 6, 7].
Non-functioning pancreatic NETs often present at an advanced stage due to mass effect or metastatic symptoms. Management involves surgical resection when feasible, along with systemic therapies such as somatostatin analogues (octreotide LAR) for tumors that express somatostatin receptors [2, 7, 9]. Clinical studies have reported a median time to tumor progression of 14.3 months in patients treated with octreotide compared to 6 months in those receiving placebo. PET/CT-based assessments have shown partial response rates of approximately 26%, stable disease in 61%, and disease progression in 11%, with a reported 5-year overall survival of nearly 61.8% among treated individuals [3-5, 9].
Peptide receptor radionuclide therapy (PRRT) has emerged as an effective option for unresectable or metastatic NETs with somatostatin receptor overexpression, significantly improving progression-free survival [10]. The NETTER-2 trial demonstrated a marked increase in median progression-free survival—22.8 months with PRRT compared to 8.5 months with placebo—highlighting its therapeutic value [11]. Given the potential for recurrence or progression, lifelong surveillance remains critical for all pancreatic NET patients [7, 8].
Conclusion
This case series highlights the clinical heterogeneity of pancreatic neuroendocrine tumors and underscores the importance of multimodal imaging, histopathological grading, and individualized therapeutic approaches. Functioning insulinomas demonstrate a high curative potential with timely surgical intervention, whereas non-functioning NETs often require systemic therapies such as somatostatin analogues and PRRT in advanced or unresectable disease, contributing to symptom control and improved quality of life. Lifelong surveillance remains essential, as systemic therapies primarily achieve tumor stabilization or reduction rather than complete cure in advanced NETs.
Acknowledgement
Departments of Pathology, Medical Gastroenterology, Surgical Gastroenterology, and Nuclear Medicine at Krishna Institute of Medical Sciences.
Conflicts of interest
Authors declare no conflicts of interest.
References
[1] Yao JC, Hassan M, Phan A. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008; 26:3063–3072.
[2] Howe JR, Cardona K, Fraker DL. The surgical management of small bowel neuroendocrine tumors: consensus guidelines of the North American Neuroendocrine Tumor Society. Pancreas. 2017; 46:715–731.
[3] Krenning EP, de Jong M, Kooij PP. Radiolabelled somatostatin analogue(s) for peptide receptor scintigraphy and radionuclide therapy. Ann Oncol. 1999; 10:23–29.
[4] Strosberg J, El–Haddad G, Wolin E. Phase 3 trial of 177Lu–dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017; 376:125–135.
[5] Brabander T, van der Zwan WA, Teunissen JJM. Long–term efficacy, survival, and safety of [177Lu–DOTA0,Tyr3]octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clin Cancer Res. 2017; 23:4617–4624.
[6] Chung A, Kwan V. Endoscopic ultrasound: an overview of its role in current clinical practice. Australas J Ultrasound Med. 2009; 12:21–29.
[7] Jensen RT, Cadiot G, Brandi ML. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology. 2012; 95:98–119.
[8] Modlin IM, Oberg K, Chung DC. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008; 9:61–72.
[9] Falconi M, Eriksson B, Kaltsas G. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non–functional pancreatic neuroendocrine tumors. Neuroendocrinology. 2016; 103:153–171.
[10] Trautwein NF, Schwenck J, Jacoby J. Long–term prognostic factors for PRRT in neuroendocrine tumors. Front Med (Lausanne). 2023; 10:1169970.
[11] Singh S, Halperin D, Myrehaug S. [177Lu]Lu–DOTA–TATE plus long–acting octreotide versus high–dose long–acting octreotide for the treatment of newly diagnosed, advanced grade 2–3, well–differentiated gastroenteropancreatic neuroendocrine tumours (NETTER–2): an open–label, randomised, phase 3 study. Lancet. 2024; 403:2807–2817.
[12] Chauhan A, Chan K, Halfdanarson TR. Critical updates in neuroendocrine tumors: Version 9 American Joint Committee on Cancer staging system for gastroenteropancreatic neuroendocrine tumors. CA Cancer J Clin. 2024; 74:359–367.
[13] Hofland J, Refardt JC, Feelders RA, Christ E, de Herder WW. Approach to the patient: insulinoma. J Clin Endocrinol Metab. 2024; 109:1109–1118.
[14] Rychlewska–Duda J, Duda. Insulinomas: comprehensive review of epidemiology, pathophysiology, clinical manifestations, diagnostic approaches, and treatment options. Qual Sport. 2025; 37:57541.