Full Text
Introduction
Alport’s syndrome is an inherited disease characterized clinically by progressive kidney disease accompanied with sensorineural hearing loss and ocular abnormalities. Alport’s syndrome accounts for 0.3 to 2.3% of end-stage kidney disease. It is related to the mutations in type IV collagen genes. Alport’s syndrome can be X-linked, autosomal recessive, or autosomal dominant [1]. Alport’s syndrome is inherited and mainly induced by a mutation within one of the collagen genes; COL4A3 and COL4A4 are located on chromosome 2 while COL4A5 is located on the X-chromosome. The X-linked form of the disease is the most common, representing nearly 80% of all AS cases, while the autosomal forms of the disease, which can be dominant or recessive depending on the gene variant, account for the remaining 20% [2].
A 30-year-old male patient with chronic kidney disease (CKD) stage 5 planned for renal transplantation presented to the ophthalmology outpatient department with complaints of progressive decrease of vision in both eyes since 2 years. After a detailed ocular examination, the patient was found to have anterior lenticonus. With further detailed history, patients found to have hearing loss observed for 2 years. The triad of lenticonus, renal failure, and hearing loss aids towards Alport’s syndrome. Further genetic analysis was done. The inheritance pattern of this gene variant was found to be autosomal recessive. Genetic analysis of parents was done. Both parents found to be carriers and his elder sibling found to have Alport's syndrome. We present the following case report of a patient with CKD stage 5 planned for renal transplantation was diagnosed with Alport's syndrome and the donor was the father of the patient who later on found to be carrier of Alport’s syndrome.
Case presentation
A 30-years-old male patient with CKD stage 5 planned for renal transplantation presented to the outpatient department of ophthalmology with a complaint of progressive decrease in vision in both eyes since 2 years. The patient has a history of frequent change in glass prescription for the past 2 years. Patient had a history of anemia and hypertension for 3 years and was on medication for the same and was recently diagnosed with end stage kidney disease. Family history- revealed second-degree parental consanguinity. Patient’s elder sibling had CKD and had undergone renal transplantation in the past. On examination, the patient looked pale, PR-116/min, BP- 150/100mmHg. On detailed ocular Examination, unaided vision was counting fingers (CF) up to 2 meters in both eyes in pinhole improvement up to 6/36 in both eyes. On slit lamp examination- lids, conjunctiva, cornea appears normal, anterior chamber was deep, quiet with round, regular, reacting pupils in both eyes. On dilation, Bilateral anterior lenticonus was seen (Figure 1) and oil droplet reflex seen in retro-illumination in both eyes (Figure 2). On dilated fundus examination, optic disc appears normal with macular foveal reflex present. On detailed history taking, patients complain of hearing loss observed for 2 years. ENT consultation was done and the patient was found to have moderate sensorineural hearing loss.
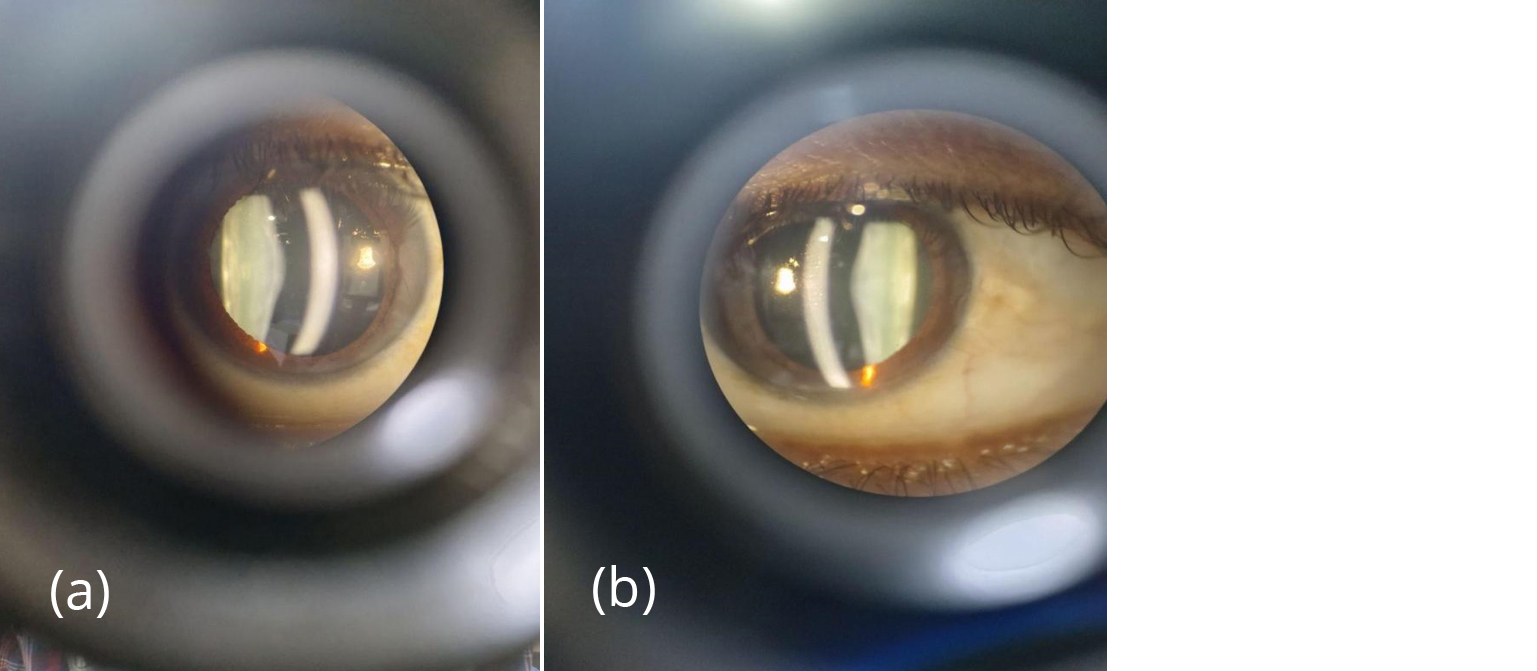
Figure 1a,b: Slit section of anterior capsule showing conical protrusion in pupillary area in both eyes.
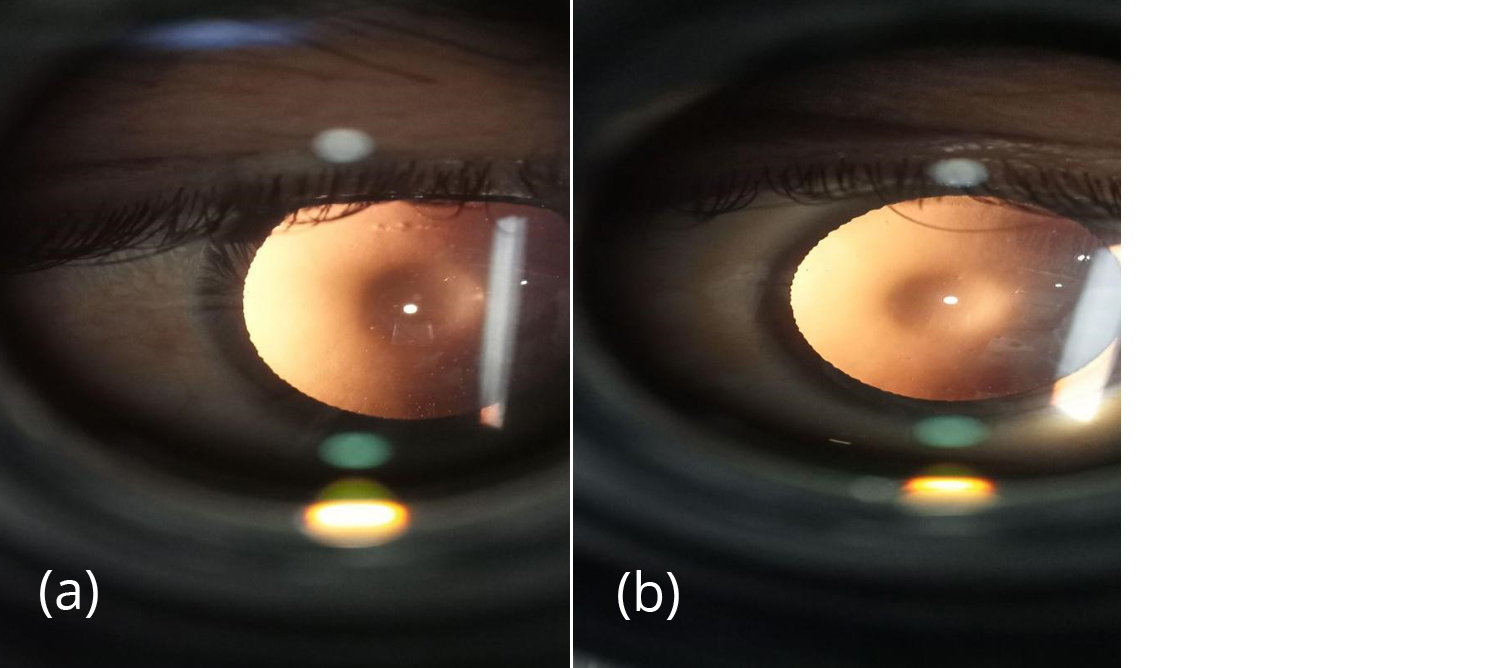
Figure 2a,b: Oil droplet reflex seen in retro-illumination.
Anterior segment OCT was suggestive of anterior lenticonus (Figure 3). SD- OCT shows temporal retinal thinning (Figure 4). Pure tone audiometry reveals bilateral sensorineural hearing loss (Figure 5). Blood investigations- hb- 6.8 gm, blood urea- 39 mg/dl, serum creatinine - 3 mg/dl, urine analysis shows hematuria and proteinuria. Ultrasound whole abdomen - bilateral grade 3 renal parenchymal disease. Based on the corroborative findings, diagnosis of bilateral anterior lenticonus of Alport’s syndrome was made. Patient was referred back to the nephrology department in view of alport's syndrome and genetic analysis was done. Genetic evaluation by NGS revealed a likely pathogenic homozygous intronic variant in COL4A3 gene. The inheritance pattern of this gene variant was autosomal recessive. Genetic analysis of parents was done. Both parents found to be carriers and his elder sibling found to have Alport's syndrome. Donor of the patient was his father initially and the decision was dropped as his father was found to have asymptomatic microscopic hematuria. Patient was referred back to the nephrology department in view of systemic condition. For visual rehabilitation, clear lens extraction with intraocular implantation was considered.

Figure 3a,b: AS-OCT shows anterior bulging.
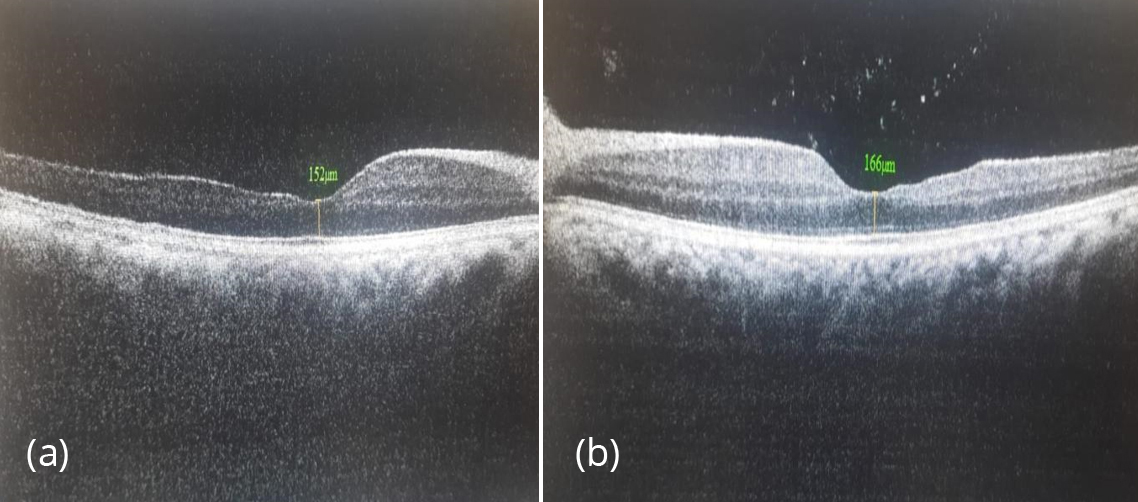
Figure 4a,b: SD-OCT shows temporal retinal thinning in both eyes.
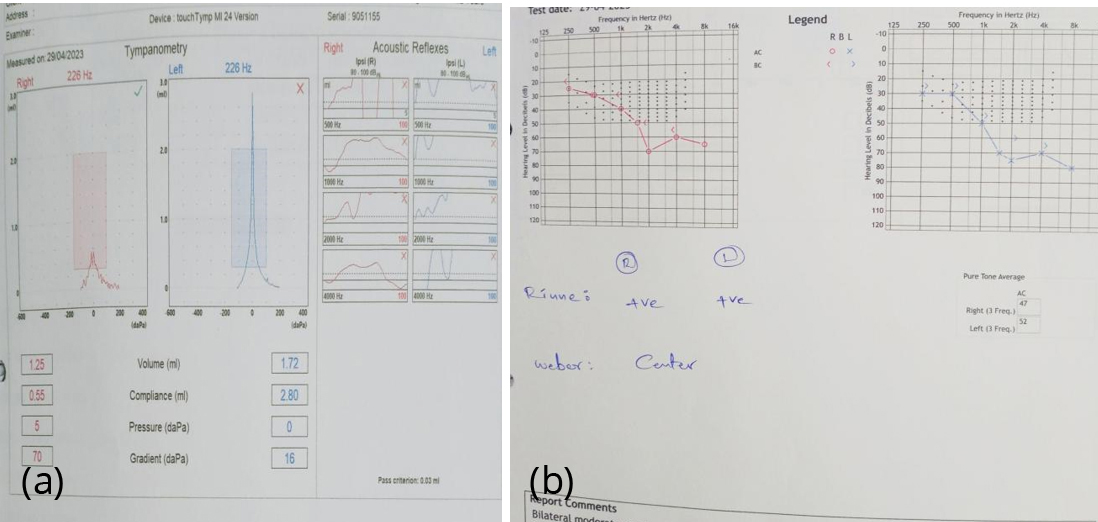
Figure 5a,b: Audiometry reveals moderate sensorineural hearing loss.
Discussion
Alport’s syndrome is a group of basement membrane abnormality due to defective type IV collagen gene and has estimated prevalence of 1 case per 5000 people and 85% of patients have the X-linked inheritance form, although there can be autosomal recessive (10%) or autosomal dominant (5%) inheritance. Men are affected more commonly and more severely.
Nephritis with haematuria secondary to basement membrane disease of the glomeruli is the most life-threatening aspect of this disorder. Progressive sensorineural hearing loss beginning with high frequencies, most pronounced at a frequency of between 2000 and 8000 Hz, which makes it difficult to distinguish speech and gives increased sensitivity to loud noises, is characteristic. The most frequent ocular findings are progressive bilateral lenticonus and it occurs in approximately 25% of patients with X-linked Alport’s syndrome and when present, it may be a pathognomonic feature [3]. Ocular abnormalities are common due to the presence of type IV collagen in the lens capsule, Descemet’s membrane, Bowman’s membrane, and retinal pigment epithelium of the eye. The characteristic ocular features of Alport’s syndrome are corneal opacities, anterior lenticonus, cataract, central peri-macular, peripheral coalescing fleck retinopathies, and temporal retinal thinning. Rarely, patients may also have posterior polymorphous corneal dystrophy and giant macular holes that impair vision [4]. The ophthalmic manifestations of Alport’s syndrome were first reported in 1954, and the characteristic triad of signs is well recognized by ophthalmologists but only visible using a slit lamp ophthalmoscope [5]. Treatment of the visual problems in these patients is an important but secondary concern due to the seriousness of the systemic disease. Clear lens extraction with intraocular lens implantation was advised for this patient but referred back to the nephrology department in view of systemic disease [6].
According to Alport’s syndrome classification, women with heterozygous variants in COL4A5 are not carriers of X-linked Alport syndrome, rather they actually have X-linked Alport syndrome and are at risk for progressive kidney disease. Similarly, individuals with heterozygous variants in COL4A3 or COL4A4 are not only carriers of autosomal recessive Alport’s syndrome but they are also at risk for progressive kidney failure. In families with documented inheritance of hereditary nephritis, living donors should be counseled with regard to possible development of the disease.
Asymptomatic males do not carry the abnormality, and heterozygous females may develop hematuria but rarely progress to renal insufficiency. Autosomal recessive inheritance occurs rarely and clinical disease expression (hematuria, progressive renal failure, and sensorineural hearing loss), is similar irrespective of the pattern of inheritance, but variable in time of appearance and severity. Screening of donors consists of examination for hematuria, renal function, auditory testing, and eye abnormalities such as anterior lenticonus, cataracts, and retinal lesions. Male relatives without hematuria can be suitable donors for patients with hereditary nephritis. Female relatives without hematuria may be considered suitable donors, however, a woman who might be a carrier should consider the possibility that she may have a child with the disease who might require transplantation [7].
Making the diagnosis of Alport’s syndrome establishes that, an individual having a familial disease that carries the risk for progression to kidney failure, should result in close monitoring of the patient and evaluation of at-risk relatives. In addition, diagnosis in adults, even those who have already advanced to kidney failure, creates the opportunity to establish the diagnosis in related adults and children who can benefit from intervention [8].
Conclusion
Timely recognition of this rare clinical condition can lessen life-threatening systemic complications. It is important to recognize Alport’s syndrome early in the course of the disease. This is facilitated by an integrated approach to diagnosis. The role of an ophthalmologist is prudent in the diagnosis of such interdisciplinary entities. Early diagnosis can improve longevity and improve the prognosis of Alport’s syndrome patients. This case scenario highlights the importance of ocular examination in patients with renal dysfunction in diagnosing the etiology and plan for appropriate management prior to renal transplantation.
Conflict of interest
Authors declare no conflict of interest.
References
[1] Hartopoa AB, Kuswadi I, Hartono H. Two Male Siblings With Alport Syndrome. World J Nephrol Urol. 2012; 1:127–129.
[2] Ramakrishnan R, Shenoy A, Meyer D. Ocular Manifestations and potential treatments of alport syndrome. J Ophthalmol. 2022: 8:9250367.
[3] Sonarkhan S, Ramappa M, Chaurasia S, Mulay K. Bilateral anterior lenticonus in a case of Alport syndrome: a clinical and histopathological correlation after successful clear lens extraction. BMJ Case Rep. 2014; 2014:bcr2013202036.
[4] Sargazi M, Dehghani S, Dahmardeh M, Mohammadi SO. Ocular manifestations of alport syndrome: report and comparison of two cases. Cureus. 2023; 15:e47373.
[5] Qasem A, Karroum R, Darwish T. A Case Report of Alport’s Syndrome: A Rare Cause of Anterior and Posterior Lenticonus. Int J Rare Dis Orph Drug. 2023; 6:1013.
[6] Aslanzadeh GA, Gharabaghi D, Naderi N. Clear lens phacoemulsification in the anterior lenticonus due to Alport Syndrome. J Med Case Rep. 2008; 2:178.
[7] Kasiske BL, Ravenscraft M, Ramos EL, Gaston RS, Bia MJ, et al. The evaluation of living renal transplant donors: clinical practice guidelines. Ad Hoc Clinical Practice Guidelines Subcommittee of the Patient Care and Education Committee of the American Society of Transplant Physicians. J Am Soc Nephrol. 1996; 7:2288–2313.
[8] Kashtan CE. Alport Syndrome: Achieving early diagnosis and treatment. Am J Kidney Dis. 2021; 77:272–279.